Capítulo 4 Proteínas
4.1 Composição de aminoácidos
seq<-"MKWVTFISLLFLFSSAYSRGVFRRDAHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEV
TEFAKTCVADESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLV
RPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELR
DEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADD
RADLAKYICENQDSISSKLKECCEKPLLEKSHCIAEVENDEMPADLPSLAADFVESKDVCKNYAEAKDVF
LGMFLYEYARRHPDYSVVLLLRLAKTYETTLEKCCAAADPHECYAKVFDEFKPLVEEPQNLIKQNCELFE
QLGEYKFQNALLVRYTKKVPQVSTPTLVEVSRNLGKVGSKCCKHPEAKRMPCAEDYLSVVLNQLCVLHEK
TPVSDRVTKCCTESLVNRRPCFSALEVDETYVPKEFNAETFTFHADICTLSEKERQIKKQTALVELVKHK
PKATKEQLKAVMDDFAAFVEKCCKADDKETCFAEEGKKLVAASQAALGL"seq<-seq[seq !="\n"]; seq # operação booleana != significa "não"## [1] "MKWVTFISLLFLFSSAYSRGVFRRDAHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEV\nTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLV\nRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELR\nDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADD\nRADLAKYICENQDSISSKLKECCEKPLLEKSHCIAEVENDEMPADLPSLAADFVESKDVCKNYAEAKDVF\nLGMFLYEYARRHPDYSVVLLLRLAKTYETTLEKCCAAADPHECYAKVFDEFKPLVEEPQNLIKQNCELFE\nQLGEYKFQNALLVRYTKKVPQVSTPTLVEVSRNLGKVGSKCCKHPEAKRMPCAEDYLSVVLNQLCVLHEK\nTPVSDRVTKCCTESLVNRRPCFSALEVDETYVPKEFNAETFTFHADICTLSEKERQIKKQTALVELVKHK\nPKATKEQLKAVMDDFAAFVEKCCKADDKETCFAEEGKKLVAASQAALGL"A seguir, obtém-se o quantitativo de uma letra específica da sequência.
library(stringr)
aa<-str_count(seq, pattern="A"); aa## [1] 63str_count contabiliza apenas a letra “A” na sequência. Dessa forma, é possível obter todos os 20 aminoácidos, repetindo-se esse comando.library(stringr)
ala<-str_count(seq, pattern="A")
arg<-str_count(seq, pattern="R")
asn<-str_count(seq, pattern="N")
asp<-str_count(seq, pattern="D")
cys<-str_count(seq, pattern="C")
glu<-str_count(seq, pattern="E")
gln<-str_count(seq, pattern="Q")
gly<-str_count(seq, pattern="G")
his<-str_count(seq, pattern="H")
ile<-str_count(seq, pattern="I")
leu<-str_count(seq, pattern="L")
lys<-str_count(seq, pattern="K")
met<-str_count(seq, pattern="M")
phe<-str_count(seq, pattern="F")
pro<-str_count(seq, pattern="P")
ser<-str_count(seq, pattern="S")
thr<-str_count(seq, pattern="T")
trp<-str_count(seq, pattern="W")
tyr<-str_count(seq, pattern="Y")
val<-str_count(seq, pattern="V")E, para visualizar o resultado numa tabela:
aa_3abrev<-c("Ala","Arg","Asn","Asp","Cys","Glu","Gln","Gly","His","Ile","Leu","Lys","Met","Phe","Pro","Ser","Thr","Trp","Tyr","Val") # vetor com os nomes de cada aminoácido
aa_quant<-c(ala,arg,asn,asp,cys,glu,gln,gly,his,ile,leu,lys,met,phe,pro,ser,thr,trp,tyr,val) # vetor com o quantitativo de aminoácidos da proteína
aa_seq<-data.frame(aa_3abrev,aa_quant) # dataframe com os resultados
colnames(aa_seq)<-c("Tipo","Qtde") # renomear as colunas
# Composição de aminoácidos em albumina de soro humano
aa_seq # apresenta a tabela## Tipo Qtde
## 1 Ala 63
## 2 Arg 27
## 3 Asn 17
## 4 Asp 36
## 5 Cys 35
## 6 Glu 62
## 7 Gln 20
## 8 Gly 13
## 9 His 16
## 10 Ile 9
## 11 Leu 64
## 12 Lys 60
## 13 Met 7
## 14 Phe 35
## 15 Pro 24
## 16 Ser 28
## 17 Thr 29
## 18 Trp 2
## 19 Tyr 19
## 20 Val 43library(knitr) # para gerar a tabela
knitr::kable(aa_seq, caption="Composição de aminoácidos em albumina de soro humano.", "pipe") # tabela| Tipo | Qtde |
|---|---|
| Ala | 63 |
| Arg | 27 |
| Asn | 17 |
| Asp | 36 |
| Cys | 35 |
| Glu | 62 |
| Gln | 20 |
| Gly | 13 |
| His | 16 |
| Ile | 9 |
| Leu | 64 |
| Lys | 60 |
| Met | 7 |
| Phe | 35 |
| Pro | 24 |
| Ser | 28 |
| Thr | 29 |
| Trp | 2 |
| Tyr | 19 |
| Val | 43 |
aa_1abrev=c("A","R","N","D","C","E","Q","G","H", "I","L","K", "M","F","P","S","T","W","Y","V")
for(i in aa_1abrev) {
aa_quant2<-str_count(seq, pattern=aa_1abrev)
return(aa_quant2) # sintaxe opcional para função com apenas uma saída
}
aa_seq<-data.frame(aa_3abrev,aa_quant2) # dataframe com os resultados
colnames(aa_seq)<-c("Tipo","Qtde") # renomear as colunas
knitr::kable(aa_seq,caption="Composição de aminoácidos em albumina de soro humano (uso de loop).", "pipe") # tabela| Tipo | Qtde |
|---|---|
| Ala | 63 |
| Arg | 27 |
| Asn | 17 |
| Asp | 36 |
| Cys | 35 |
| Glu | 62 |
| Gln | 20 |
| Gly | 13 |
| His | 16 |
| Ile | 9 |
| Leu | 64 |
| Lys | 60 |
| Met | 7 |
| Phe | 35 |
| Pro | 24 |
| Ser | 28 |
| Thr | 29 |
| Trp | 2 |
| Tyr | 19 |
| Val | 43 |
str_count retém em si um loop interno, já que aplica uma função de contagem de elementos a uma sequência, a partir de um padrão pré-definido (o vetor aa_1abrev, no caso). Dessa forma, pode-se simplificar ainda mais o script, não necessitando do loop externo.str_count(seq, pattern=aa_1abrev)## [1] 63 27 17 36 35 62 20 13 16 9 64 60 7 35 24 28 29 2 19 43y=c(1,2,4,8,16,32)
mean(y)## [1] 10.5sum(y)## [1] 63# Tamanho médio estimado de uma proteína a partir do no. de resíduos de aminoácidos
prot.tamanho<-function(x){
MM<-x*110 # 'x' representa o número de aminoácidos da proteína
return(MM)
}
prot.tamanho(575) # no. de resíduos de aminoácidos de albumina humana## [1] 63250apply, composta pelos comandos apply, sapply, tapply, lapply, e mapply. Embora possuam processamento mais rápido que funções de loop externo para uso de matrizes muito complexas, cada qual é voltado a um objeto distinto ou situação específica do R (retorno de lista, vetor ou matriz), permite o uso de subset (subconjuntos de dados), utiliza funções do R ou funções previamente definidas pelo usuário, e roda em apenas uma linha de comando. Essas vantagens contrapõe-se ao uso de loop for aplicado para vetores. Contudo, a vetorização opera muito bem quando se deseja aplicar ou mapear uma função a um vetor/matriz/lista. Quando, por outro lado, se deseja aplicar uma função cujo resultado dependa de mais de um vetor/matriz/lista, o loop for torna-se indispensável, como na titulação de ácidos fracos do capítulo 3.aa_ac<-aa_seq[4,2]+aa_seq[6,2] # AA ácicos
aa_bas<-aa_seq[2,2]+aa_seq[9,2]+aa_seq[12,2] # AA básicos
aa_arom<-aa_seq[14,2]+aa_seq[18,2]+aa_seq[19,2] # AA aromáticos
aa_alif<-aa_seq[10,2]+aa_seq[11,2]+aa_seq[15,2]+aa_seq[1,2]+aa_seq[20,2] # AA alifáticos
aa_pol<-aa_seq[3,2]+aa_seq[5,2]+aa_seq[7,2]+aa_seq[8,2]+aa_seq[13,2]+aa_seq[16,2]+aa_seq[17,2] ## AA polares neutrosaa_tot<-str_count(seq,pattern="") # comprimento da sequência
class_perc<-round(c(aa_ac,aa_bas,aa_arom,aa_alif,aa_pol)/aa_tot*100)E agora, sim, constroi-se a tabela.
aa_class<-c("ácido","básico","aromático","alifático","polar")
aa_perc<-data.frame(aa_class,class_perc) # dataframe com os resultados
colnames(aa_perc)<-c("Classe","%") # renomear as colunas
knitr::kable(aa_perc, caption="Distribuição de classes de aminoácidos em albumina humana.", "pipe") # tabela| Classe | % |
|---|---|
| ácido | 16 |
| básico | 17 |
| aromático | 9 |
| alifático | 33 |
| polar | 24 |
4.2 Tabela de Purificação de Proteínas & R como planilha eletrônica
Para exemplificar a construção de uma planilha simples, tomemos como exemplo uma Tabela de Purificação de Proteínas, usualmente utilizada em Biotecnologia e áreas afins. A forma mais simples de construção de uma planilha envolve 1) a elaboração individual de vetores, e 2) a união dos vetores em uma planilha.
Os procedimentos para purificação (ou isolamento, fracionamento) proteica envolvem técnicas como tratamento químico (precipitação por sulfato de amônio, acetona), tratamento ácido, tratamento térmico, diálise, cromatografia (filtração molecular, troca-iônica, afinidade, fase reversa), entre outros. Para aferição do grau de pureza da amostra obtida utilizam-se normalmente a eletroforese simples, focalização isoelétrica, eletroforese 2D, uso de anticorpos monoclonais, e ensaios de atividade específicos, dentre vários.
Para a tabela de purificação são exigidos somente os vetores de massa de amostra e de atividade enzimática da amostra, obtidos em cada etapa de purificação. Uma planilha simples poderia ser construida como:
# Elaboração de planilha simples de purificação de enzima
# (cada elemento do vetor representa uma etapa de purificação)
# 1. Definição dos vetores principais:
prot.total <- c(6344,302,145,34,10,3.8) # proteína, mg
ativ.tot <- c(200,122,106,70,53,24)*1000 # atividade, U
# 2. Construção da planilha:
purif.plan <- data.frame(prot.total,ativ.tot); purif.plan## prot.total ativ.tot
## 1 6344.0 200000
## 2 302.0 122000
## 3 145.0 106000
## 4 34.0 70000
## 5 10.0 53000
## 6 3.8 24000purif.plan2 <- cbind(prot.total,ativ.tot); purif.plan2## prot.total ativ.tot
## [1,] 6344.0 200000
## [2,] 302.0 122000
## [3,] 145.0 106000
## [4,] 34.0 70000
## [5,] 10.0 53000
## [6,] 3.8 24000# Edição de nome de colunas
colnames(purif.plan2) <-c("totalProt","enzAtiv")
purif.plan2## totalProt enzAtiv
## [1,] 6344.0 200000
## [2,] 302.0 122000
## [3,] 145.0 106000
## [4,] 34.0 70000
## [5,] 10.0 53000
## [6,] 3.8 24000purif.plan3 <-data.frame(prot.total,ativ.tot,ativ.tot/prot.total)
options(digits=1) # opção para no. de casas decimais
colnames(purif.plan3)<-c("prot.total","ativ.tot","ativ.specif")
rownames(purif.plan3) <-c("extr.bruto","NH4SO2","acetona","Sephadex G-100","DEAE-celulose","C8-fase rev")
purif.plan3 ## prot.total ativ.tot ativ.specif
## extr.bruto 6344 2e+05 32
## NH4SO2 302 1e+05 404
## acetona 145 1e+05 731
## Sephadex G-100 34 7e+04 2059
## DEAE-celulose 10 5e+04 5300
## C8-fase rev 4 2e+04 6316# Edição simples de planilha (alterações de valores e nomes de colunas)
purif.plan4 <- edit(purif.plan3) # ou data.entry( )# Importação de dados de outra planilha (CSV):
# 1. Importação com nome da planilha desejada:
purif.plan5 <- read.table("planilha.csv", header = T, sep = ",")
# 2. Importação com tela de busca da planilha desejada:
purif.plan5 <- frame <-read.csv( file.choose( ) )library(tibble)
purif.plan6 <- as_tibble(purif.plan3); purif.plan6## # A tibble: 6 × 3
## prot.total ativ.tot ativ.specif
## <dbl> <dbl> <dbl>
## 1 6344 200000 31.5
## 2 302 122000 404.
## 3 145 106000 731.
## 4 34 70000 2059.
## 5 10 53000 5300
## 6 3.8 24000 6316.# Tabela de purificação de enzima com pacote 'dplyr':
library(dplyr)##
## Attaching package: 'dplyr'## The following object is masked from 'package:seqinr':
##
## count## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, unionpurif.plan7 <- mutate(purif.plan,ativ.esp=ativ.tot/prot.total)
purif.plan7## prot.total ativ.tot ativ.esp
## 1 6344 2e+05 32
## 2 302 1e+05 404
## 3 145 1e+05 731
## 4 34 7e+04 2059
## 5 10 5e+04 5300
## 6 4 2e+04 6316ativ.tot.kU <- transmute(purif.plan7, ativ.tot=ativ.tot/1e3)
ativ.tot.kU # vetor de atividade específica em U x 10^3## ativ.tot
## 1 200
## 2 122
## 3 106
## 4 70
## 5 53
## 6 24purif.plan8 <- mutate(purif.plan7,
purif=ativ.esp/ativ.esp[1], # nível de purificação
rend.perc = 100*ativ.tot/ativ.tot[1]) # rendimento percentual
# Convetendo à tabela...
library(knitr)
knitr::kable(purif.plan8, caption="Tabela de purificação para uma enzima", "pipe")| prot.total | ativ.tot | ativ.esp | purif | rend.perc |
|---|---|---|---|---|
| 6344 | 2e+05 | 32 | 1 | 100 |
| 302 | 1e+05 | 404 | 13 | 61 |
| 145 | 1e+05 | 731 | 23 | 53 |
| 34 | 7e+04 | 2059 | 65 | 35 |
| 10 | 5e+04 | 5300 | 168 | 26 |
| 4 | 2e+04 | 6316 | 200 | 12 |
library(DT)
purif.plan9 <-as.data.frame(purif.plan8)
rownames(purif.plan9) <-c("extr.bruto","NH4SO2","acetona","Sephadex G-100","DEAE-celulose","C8-fase rev") # converte a tabela de purificação em planilha para se utilizada pelo pacote DT
datatable(purif.plan9) %>% formatRound(1:5,1) # colunas com 1 casa decimalDT::datatable(purif.plan9, editable = 'cell')4.3 Interação de oxigênio com mioglobina e hemoglobina
\[\begin{equation} y=\frac{pO_2}{K_{50}+pO_2} \tag{4.1} \end{equation}\]
Por outro lado, o valor de \(K_{50}\) para a hemoglobina é de 26 mmHg, mas sua função exprime-se de forma diferente à da mioglobina:
\[\begin{equation} y=\frac{pO_2^{nH}} {K_{50}^{nH}+pO_2^{nH}} \tag{4.2} \end{equation}\]
Nessa equação (4.2), nH representa o coeficiente de cooperatividade de Hill, que resume a energia distribuida entre as quatro constantes microscópicas de dissociação de O\(_{2}\) aos quatro centros porfirínicos da hemoglobina (grupos heme). Simulando ambas as curvas:
K50=2.8
curve(x/(K50+x),xlim=c(0,100),
xlab="pO2 (mmHg)", ylab="y",lty="dotted")
K50=26
nH=2.8
curve(x^nH/(K50^nH+x^nH),xlim=c(0,100),
xlab="pO2 (mmHg)", ylab="y",col="red",
add=TRUE) # "add" permite adicionar curvas ao gráfico
abline(0.5,0, lty=2) # acrescenta linha de base em meia saturação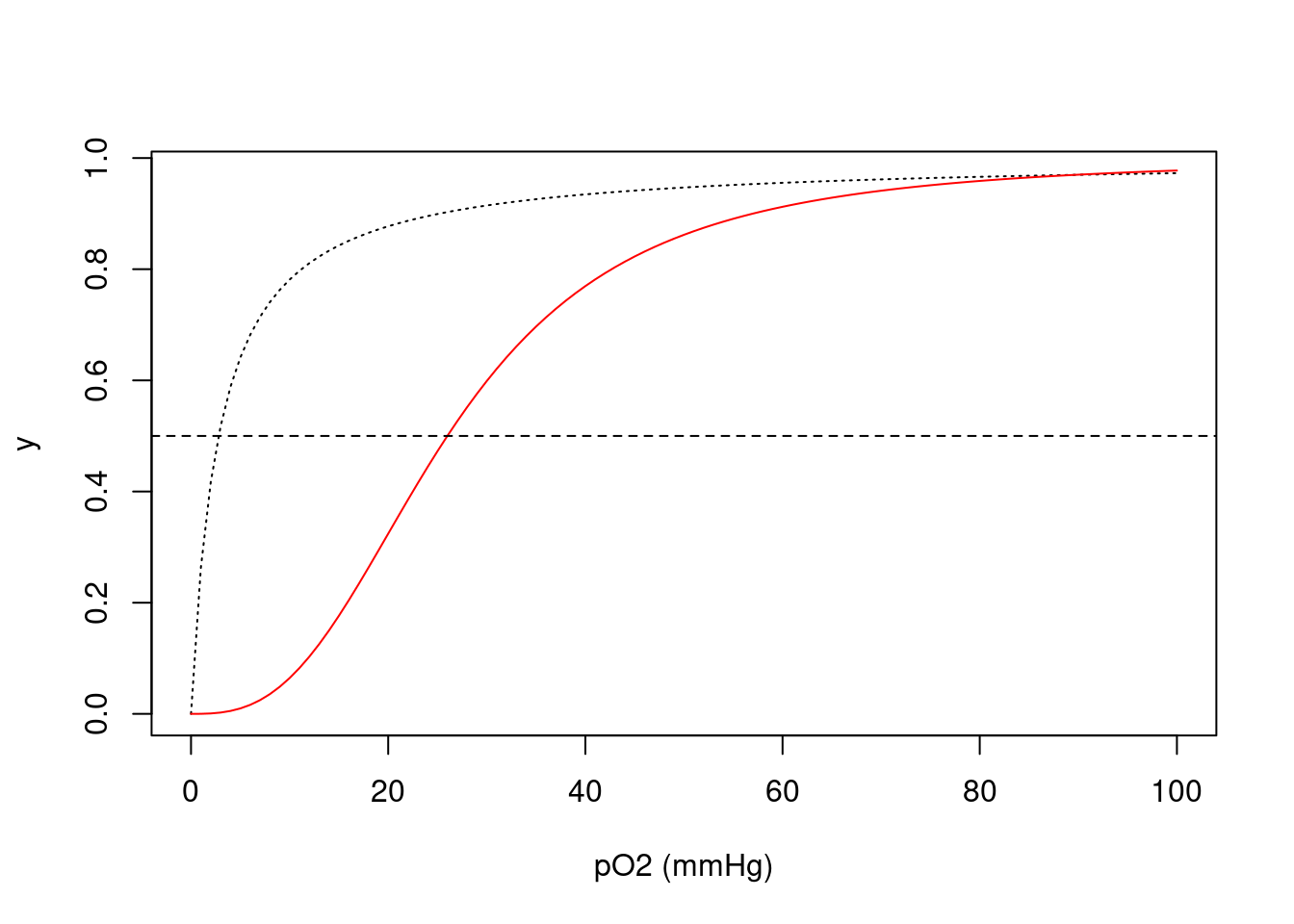
Figura 4.1: Isoterma de saturação de oxigênio à mioglobina (linha contínua) e hemoglobina (linha pontilhada), indicando o intercepto em pO2 de 50% (meia saturação).
\[\begin{equation} y=\frac{K1*L+2*K2*K1*L^2+3*K3*K2*K1*L^3+4*K4*K3*K2*K1*L^4} {4*(1+K1*L+2*K2*K1*L^2+3*K3*K2*K1*L^3+4*K4*K3*K2*K1*L^4)} \tag{4.3} \end{equation}\]
\[\begin{equation} Ki_{corr} = \frac{i}{N-1+i}*Ki \tag{4.4} \end{equation}\]
K=c(0.011,0.016,0.118,0.400) # vetor de constantes microscópicas de dissociação de Hb para O2
L<-seq(1,201,2) # vetor de teores de O2
Kcorr = c() # inicializa um vetor vazio para saída do vetor corrigido de Ki
N = 4 # declara o número de sítios na Hb
for (i in 1:N) Kcorr[i] = i/(N-i+1)*K[i]
Kcorr # apresenta o vetor de valores de Ki corrigidos para o efeito estatístico## [1] 0.003 0.011 0.177 1.600numer<-K[1]*L+2*K[2]*K[1]*L^2+3*K[3]*K[2]*K[1]*L^3+4*K[4]*K[3]*K[2]*K[1]*L^4
denom<-1+numer
y=numer/denom
plot(L,y,xlab="pO2",type="l",col=2)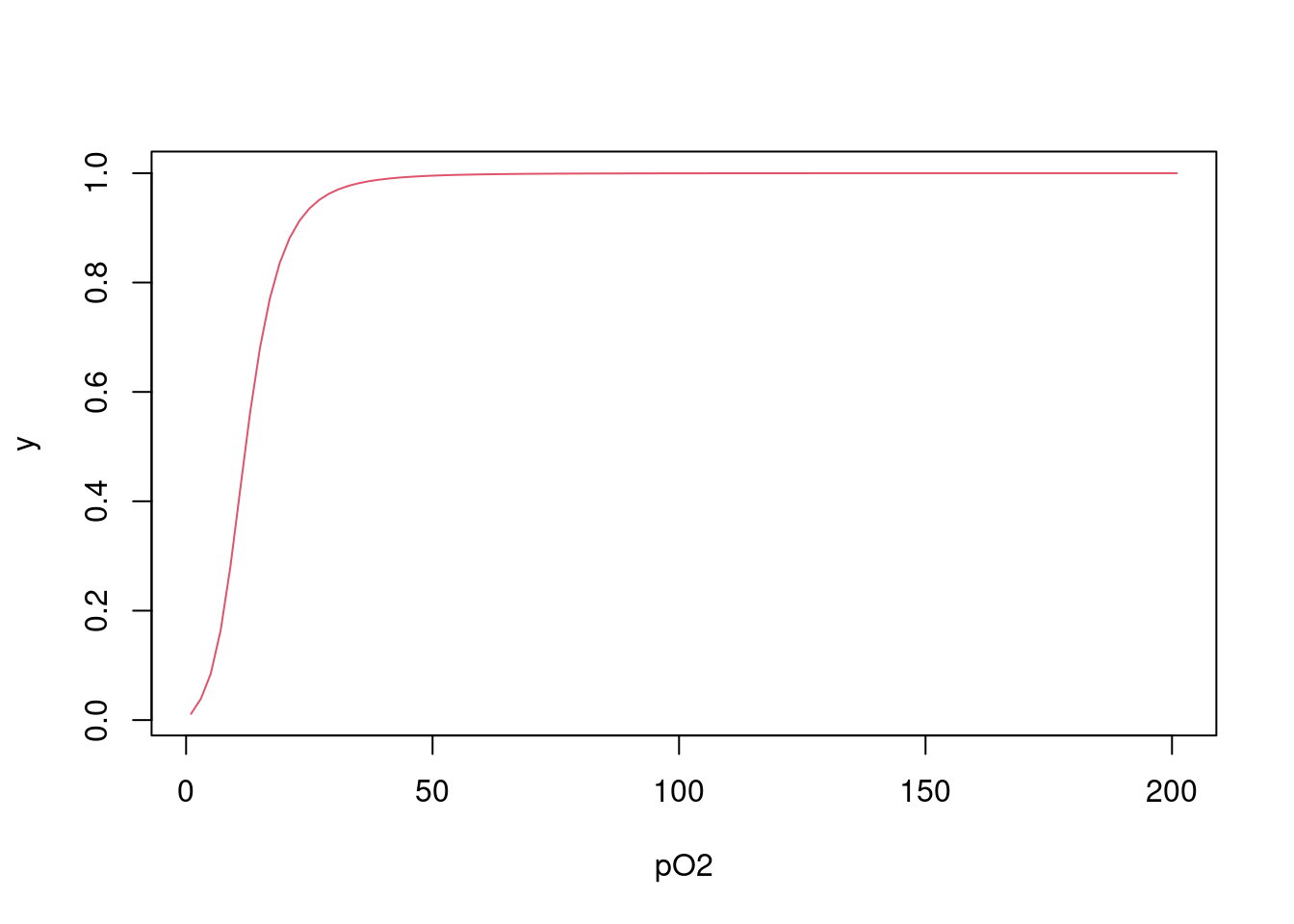
Figura 4.2: Isoterma de saturação de oxigênio à hemoglobina pela equação de Adair.
# Cálculo de y em cada L
Yi = function(L,Kcorr) {
N = length(Kcorr)
conc = c()
conc[1] = L*Kcorr[1]
for (i in 2:N) conc[i] = conc[i-1]*L*Kcorr[i]
numer2 = sum((1:N)*conc)/N
denom2 = 1 + sum(conc)
return(numer2/denom2)
}
# Cálculo de y para o vetor de L
Y = function(L,Kcorr) {
YY= c()
for (j in 1:length(L)) YY[j] = Yi(L[j],Kcorr)
return(YY)
}
# Aplicação da função de y para L e gráfico
Yfinal = Y(L,Kcorr)
plot(L,Yfinal,type="l", col=2, xlab = "pO2", ylab = "y")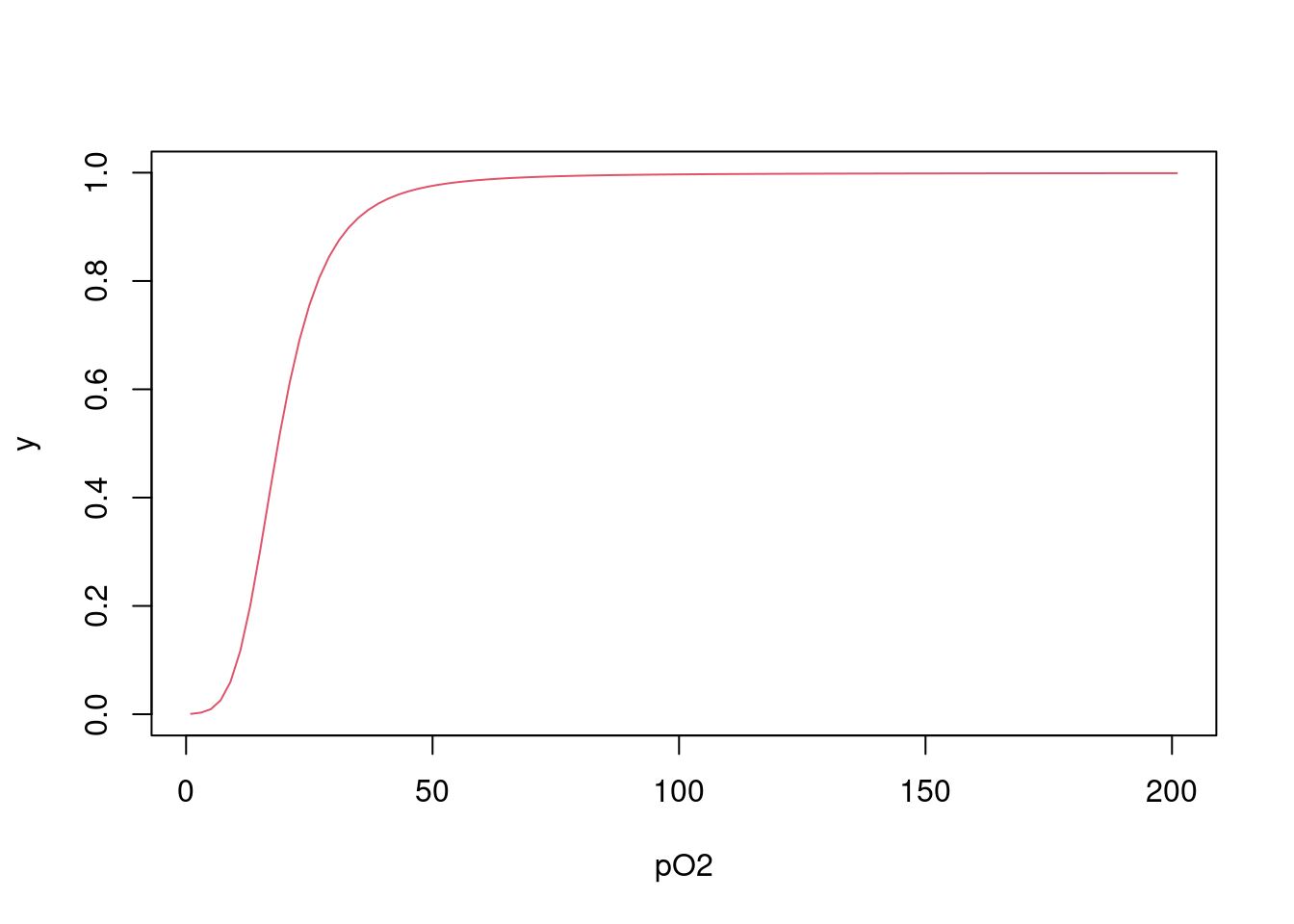
Figura 4.3: Curva de saturação de oxigênio à hemoglobina obtida por iteração da equação de Adair, tal como corrigida para o efeito estatístico.
4.4 Alguns pacotes do R para estudo de proteínas
seqinr vista no capítulo 3, e que computa diversos valores e informações para sequências proteicas, tais como pI, index de hidroxipatia, distribuição de resíduos, entre outros. O sítio do projeto 7 contém informação detalhada para seu uso. Utilizando-se o mesmo procedimento para obtenção da sequência FASTA para a lisozima do capítulo 3 (código CAA32175 no sítio NCBI), pode-se obter um conjunto extenso de informações da proteína, como exemplificado abaixo:library(seqinr)
lisozima<-c("KVFERCELARTLKRLGMDGYRGISLANWMCLAKWESGYNTRATNYNAGDRSTDYGIFQINSRYWCNDGKT
PGAVNACHLSCSALLQDNIADAVACAKRVVRDPQGIRAWVAWRNRCQNRDVRQYVQGCGV")
seq_liso<-s2c(lisozima) # converte sequência de string de aminoácidos para o padrão do seqinr (vetor de caracteres)
seq_liso2<-seq_liso[seq_liso !="\n"] # eliminação de espaços exigida pelo seqinr advindos do procedimento de copiar/colar.
seq_liso2## [1] "K" "V" "F" "E" "R" "C" "E" "L" "A" "R" "T" "L" "K" "R" "L" "G" "M" "D"
## [19] "G" "Y" "R" "G" "I" "S" "L" "A" "N" "W" "M" "C" "L" "A" "K" "W" "E" "S"
## [37] "G" "Y" "N" "T" "R" "A" "T" "N" "Y" "N" "A" "G" "D" "R" "S" "T" "D" "Y"
## [55] "G" "I" "F" "Q" "I" "N" "S" "R" "Y" "W" "C" "N" "D" "G" "K" "T" "P" "G"
## [73] "A" "V" "N" "A" "C" "H" "L" "S" "C" "S" "A" "L" "L" "Q" "D" "N" "I" "A"
## [91] "D" "A" "V" "A" "C" "A" "K" "R" "V" "V" "R" "D" "P" "Q" "G" "I" "R" "A"
## [109] "W" "V" "A" "W" "R" "N" "R" "C" "Q" "N" "R" "D" "V" "R" "Q" "Y" "V" "Q"
## [127] "G" "C" "G" "V"pmw(seq_liso2) # peso molecular da proteína## [1] 14701aaa(seq_liso2) # distribuição de resíduos## [1] "Lys" "Val" "Phe" "Glu" "Arg" "Cys" "Glu" "Leu" "Ala" "Arg" "Thr" "Leu"
## [13] "Lys" "Arg" "Leu" "Gly" "Met" "Asp" "Gly" "Tyr" "Arg" "Gly" "Ile" "Ser"
## [25] "Leu" "Ala" "Asn" "Trp" "Met" "Cys" "Leu" "Ala" "Lys" "Trp" "Glu" "Ser"
## [37] "Gly" "Tyr" "Asn" "Thr" "Arg" "Ala" "Thr" "Asn" "Tyr" "Asn" "Ala" "Gly"
## [49] "Asp" "Arg" "Ser" "Thr" "Asp" "Tyr" "Gly" "Ile" "Phe" "Gln" "Ile" "Asn"
## [61] "Ser" "Arg" "Tyr" "Trp" "Cys" "Asn" "Asp" "Gly" "Lys" "Thr" "Pro" "Gly"
## [73] "Ala" "Val" "Asn" "Ala" "Cys" "His" "Leu" "Ser" "Cys" "Ser" "Ala" "Leu"
## [85] "Leu" "Gln" "Asp" "Asn" "Ile" "Ala" "Asp" "Ala" "Val" "Ala" "Cys" "Ala"
## [97] "Lys" "Arg" "Val" "Val" "Arg" "Asp" "Pro" "Gln" "Gly" "Ile" "Arg" "Ala"
## [109] "Trp" "Val" "Ala" "Trp" "Arg" "Asn" "Arg" "Cys" "Gln" "Asn" "Arg" "Asp"
## [121] "Val" "Arg" "Gln" "Tyr" "Val" "Gln" "Gly" "Cys" "Gly" "Val"AAstat(seq_liso2,plot=TRUE) # gráfico de distribuição, composição e proporção de resíduos, valor de pI 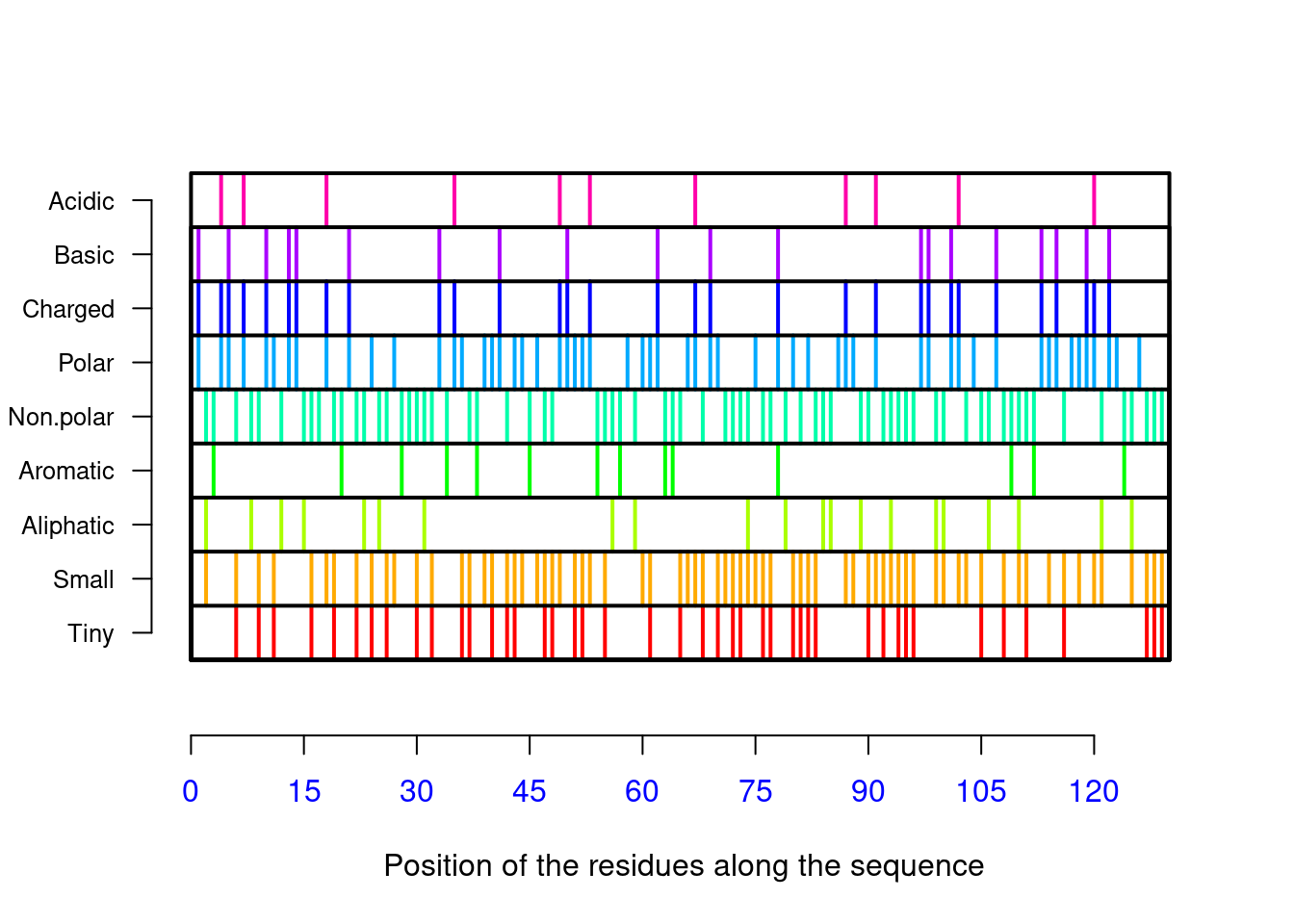
## $Compo
##
## * A C D E F G H I K L M N P Q R S T V W Y
## 0 14 8 8 3 2 11 1 5 5 8 2 10 2 6 14 6 5 9 5 6
##
## $Prop
## $Prop$Tiny
## [1] 0.3
##
## $Prop$Small
## [1] 0.6
##
## $Prop$Aliphatic
## [1] 0.2
##
## $Prop$Aromatic
## [1] 0.1
##
## $Prop$Non.polar
## [1] 0.6
##
## $Prop$Polar
## [1] 0.4
##
## $Prop$Charged
## [1] 0.2
##
## $Prop$Basic
## [1] 0.2
##
## $Prop$Acidic
## [1] 0.08
##
##
## $Pi
## [1] 9seqinr inclui-se a conversão de aminoácidos para abreviações de 1 e 3 letras (ae aaa, respectivamente), listagem de 544 propriedades físico-químicas dos 20 aminoácidos proteicos (aaindex),pK (autoexplicativo, e visto anteriormente), e cômputo isolado de pI (computePI) e de massa molecular (pmw), além de várias outras, tanto para proteômica como para genômica.Outro pacote do R interessante para estudo de proteínas é o Peptides 8, que também computa diversas propriedades físico-químicas para sequências de aminoácidos, além de possibilitar a integração de plotagem com o pacote de dinâmica molecular GROMACS. Como para o
seqinr, o Peptides necessita de conversão da sequência em string para o padrão vetorial reconhecido. Entre as funções do pacote destacam-se o cômputo de 66 descritores para cada aminoácido de uma sequência (aaDescriptors), a composição da sequência por classificação dos resíduos (aaComp), o cômputo de índice alifático (aIndex), o índice de hidrofobicidade (hydrophobicity), índice de instabilidade (instalIndex), relação de massa/carga (mz), massa molecular (mw), e pI (pI), entre outros.Entre pacotes mais direcionados ao estudo comparativo e visualização de estruturas, bem como para descritores de bioinformática e quimiogenômica vale mencionar
Bio3d, Autoplotprotein, protr, BioMedR, e UniprotR, entre muitos.References
Algumas práticas de programação (Best Codes): 1) organizar um projeto em pastas (ex: dados,figuras,scripts) ou criar um pacote do R como opção; 2) criar seções num código pra facilitar localização; 3) nomear os code chunks (pedaços de código); 4) colocar no início do código as bibliotecas utilizadas, fontes, e chamada de dados (evita procurar algo necessário pro script rodar ao longo do código); 5) identar, preferivelmente com 1 ou 2 comandos por linha; 6) parâmetros de função sempre dentro de função; 7) evitar parâmetros globais; 8) não usar ‘attach’; 8) usar parâmetros com nomes intuitivos (e não x e y; ex: nome_função); 9) atribuir nomes à objetos com uma das três convenções nominais (ex: KiCompet, ki_compet, ki.compet).↩︎
Sítio do projeto Seqinr: http://seqinr.r-forge.r-project.org/↩︎
Pacote Peptides: https://cran.r-project.org/web/packages/Peptides/index.html↩︎