Capítulo 6 Interação Bimolecular
O formalismo mais comum para interação biomolecular é o que envolve a formação de complexo adsortivo entre uma proteína e um ligante (ligand binding), exemplificado para íons (Ca, Mg, etc), fármacos e candidatos, produtos naturais, e antígenos, dentre vários.
Perguntas simples acerca da interação ligante-proteína podem elucidar diversas características da formação de tais complexos, como:
- Quanto de proteína/ligante estão presentes ?
- Quanto do complexo é formado ?
- Quão rápido o complexo associa/dissocia ?
- Quais os mecanismos envolvidos ?
Onde P representa o teor de proteína livre, L o ligante livre, e PL o complexo formado. As taxas de reação são definidas para a formação (k; Ms) e dissociação (k; s) do complexo.
Onde Kd representa a constante de equilíbrio de dissociação para o complexo PL formado, tal como condicionado ao equilíbrio de formação/dissociação do complexo (v = v), e também definido como:
6.1 Modelos de Interação e Representações Lineares
Mantida essa similaridade com o formalismo da equação de Michaelis-Menten, da mesma maneira decorrem as linearizações para a Eq. (6.4), bem como ajustes não lineares à mesma, na busca de uma solução analítica para os parâmetros termodinâmicos Kd e n. Exemplificando um trecho de código para as linearizações mais comuns no tratamento de dados de interação ligante-proteína:
L=c(0.1,0.2,0.5,1,5,10,20)*1e-6
Kd=1e-6;n=1
v=n*L/(Kd+L)
par(mfrow=c(2,3)) # estabelece área de plot pra 6 gráficos
plot(L,v,type="o",main="Direto")
plot(log(L),v,type="o",main="Langmuir")
plot(1/L,1/v,type="o",main="Klotz")
plot(v,v/L,type="o",main="Scatchard")
plot(L,L/v,type="o",main="Woolf")
plot(log10(L),log10(v/(n-v)),type="o",main="Hill") 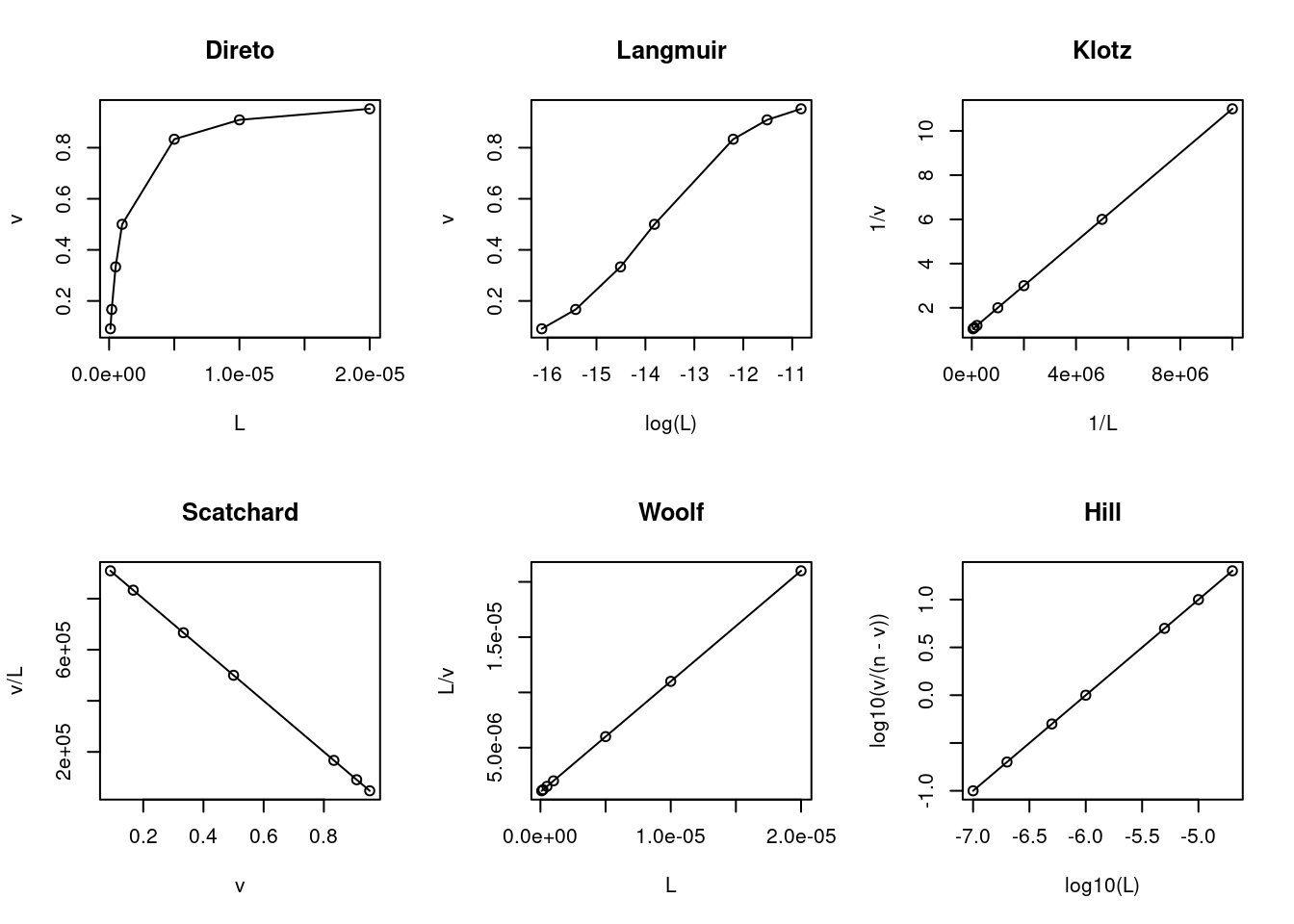
Figura 6.1: Principais linearizações da isoterma de ligação ligante-proteína.
par(mfrow=c(1,1)) # volta à janela gráfica normalL=c(0.1,0.2,0.5,1,5,10,20)*1e-6
Kd1=2e-6;n1=1;
Kd2=2e-8;n2=1
v=(n1*L/(Kd1+L))+(n2*L/(Kd2+L))
par(mfrow=c(2,3)) # estabelece área de plot pra 6 gráficos
plot(L,v,type="o",main="Direto")
plot(log(L),v,type="o",main="Langmuir")
plot(1/L,1/v,type="o",main="Klotz")
plot(v,v/L,type="o",main="Scatchard")
plot(L,L/v,type="o",main="Woolf")
plot(log10(L),log10(v/(n1+n2-v)),type="o",main="Hill") # n1+n2=ntot no Hill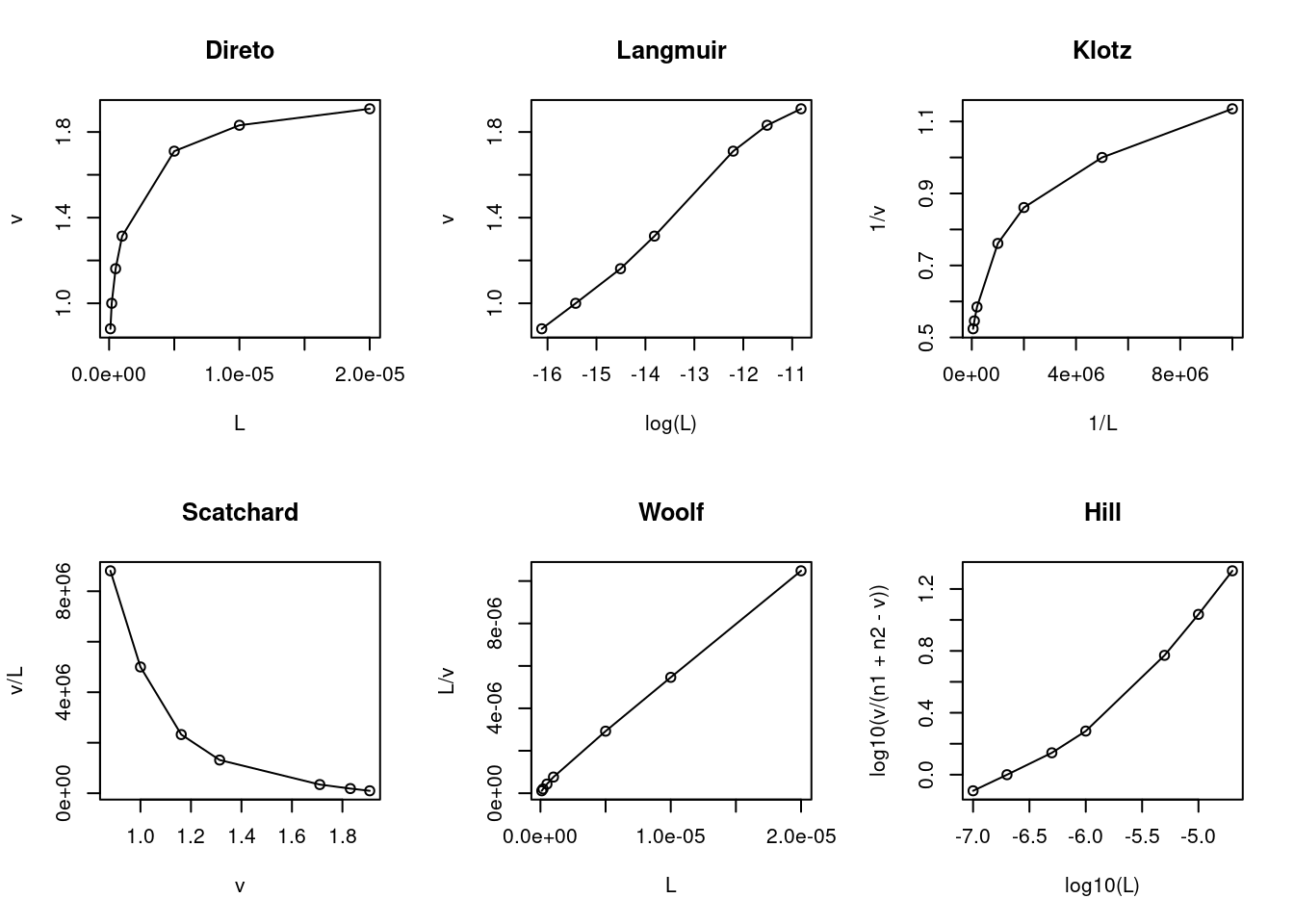
Figura 6.2: Modelo e linearizações para heterogeneidade de 2 conjuntos de sítios de ligação
par(mfrow=c(1,1)) # volta à janela gráfica normalL=c(0.1,0.2,0.5,1,5,10,20)*1e-6
Kd1=2e-6;n1=1
Kd2=2e-5;n2=1
nH=0.5
v=(n1*L*1/Kd1)/(1+1/Kd1*L)+((n2*1/Kd1*1/Kd2*L^2)/(1+1/Kd1*L)*(1+1/Kd2*L))
par(mfrow=c(2,3)) # estabelece área de plot pra 6 gráficos
plot(L,v,type="o",main="Direto")
plot(log(L),v,type="o",main="Langmuir")
plot(1/L,1/v,type="o",main="Klotz")
plot(v,v/L,type="o",main="Scatchard")
plot(L,L/v,type="o",main="Woolf")
plot(log10(L),log10(v/(n-v)),type="o",main="Hill") # n1+n2=ntot no Hill## Warning in xy.coords(x, y, xlabel, ylabel, log): NaNs produced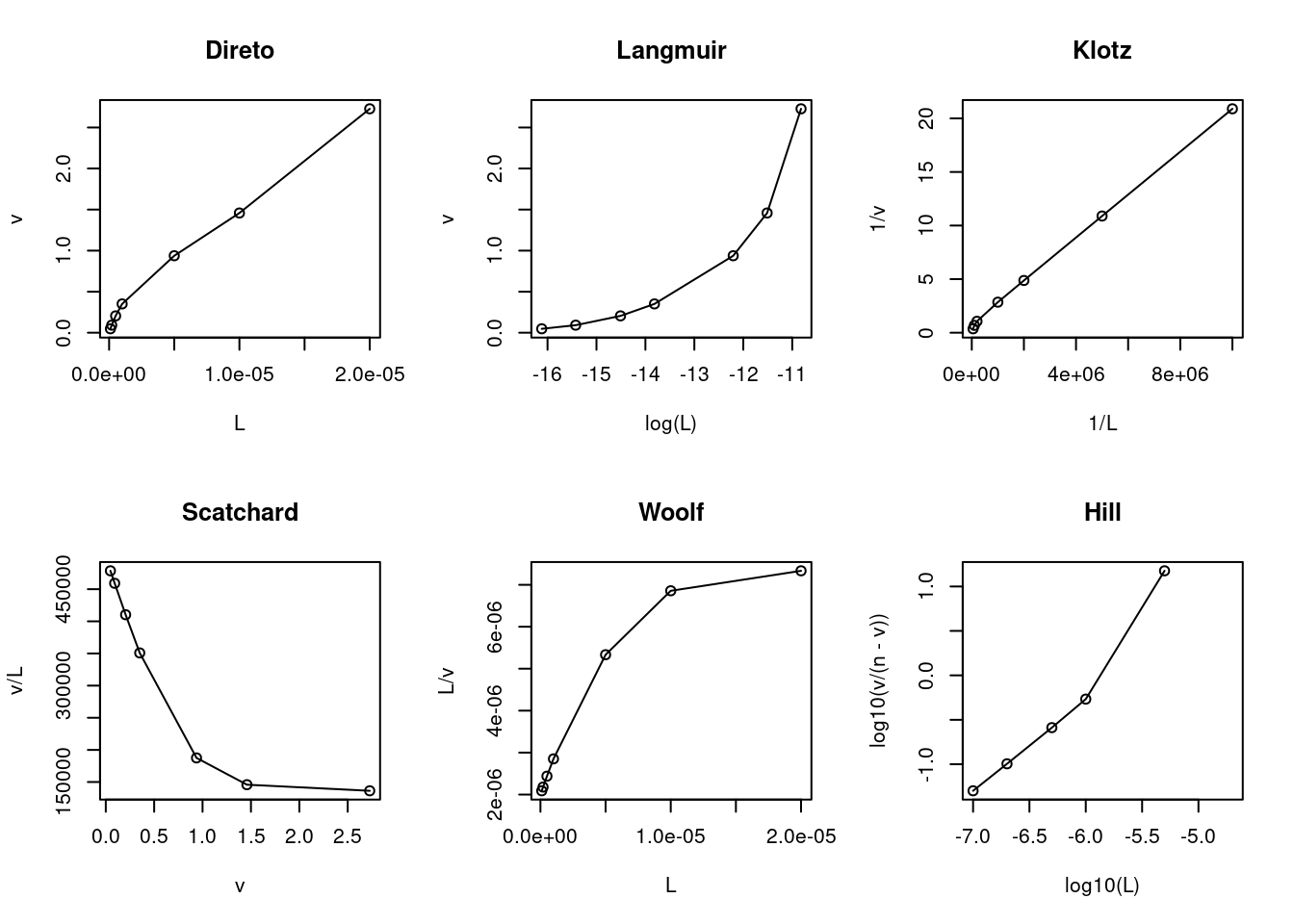
Figura 6.3: Modelo e linearizações para criação de novo sítio: 1-site creator.
par(mfrow=c(1,1)) # volta à janela gráfica normalL=c(0.1,0.2,0.5,1,5,10,20)*1e-6
Kd=2e-6;n=1
nH=0.5
v=(n*L^nH/(Kd+L^nH))
par(mfrow=c(2,3)) # estabelece área de plot pra 6 gráficos
plot(L,v,type="o",main="Direto")
plot(log(L),v,type="o",main="Langmuir")
plot(1/L,1/v,type="o",main="Klotz")
plot(v,v/L,type="o",main="Scatchard")
plot(L,L/v,type="o",main="Woolf")
plot(log10(L),log10(v/(n-v)),type="o",main="Hill") # n1+n2=ntot no Hill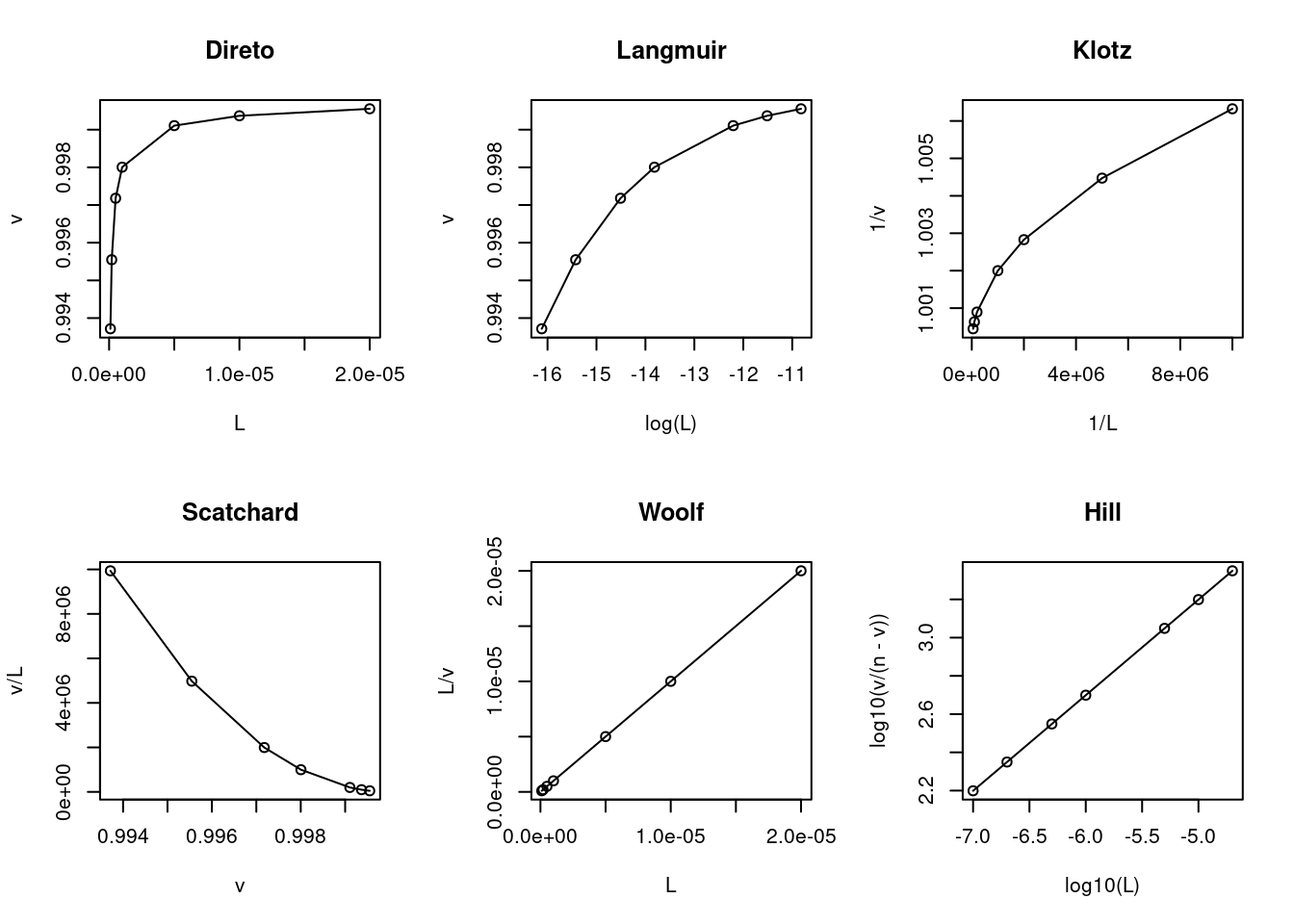
Figura 6.4: Modelo e linearizações para cooperatividade negativa de sítios de ligação.
par(mfrow=c(1,1)) # volta à janela gráfica normalL=c(0.1,0.2,0.5,1,5,10,20)*1e-6
Kd=2e-6;n=1
nH=1.5
v=(n*L^nH/(Kd+L^nH))
par(mfrow=c(2,3)) # estabelece área de plot pra 6 gráficos
plot(L,v,type="o",main="Direto")
plot(log(L),v,type="o",main="Langmuir")
plot(1/L,1/v,type="o",main="Klotz")
plot(v,v/L,type="o",main="Scatchard")
plot(L,L/v,type="o",main="Woolf")
plot(log10(L),log10(v/(n-v)),type="o",main="Hill") # n1+n2=ntot no Hill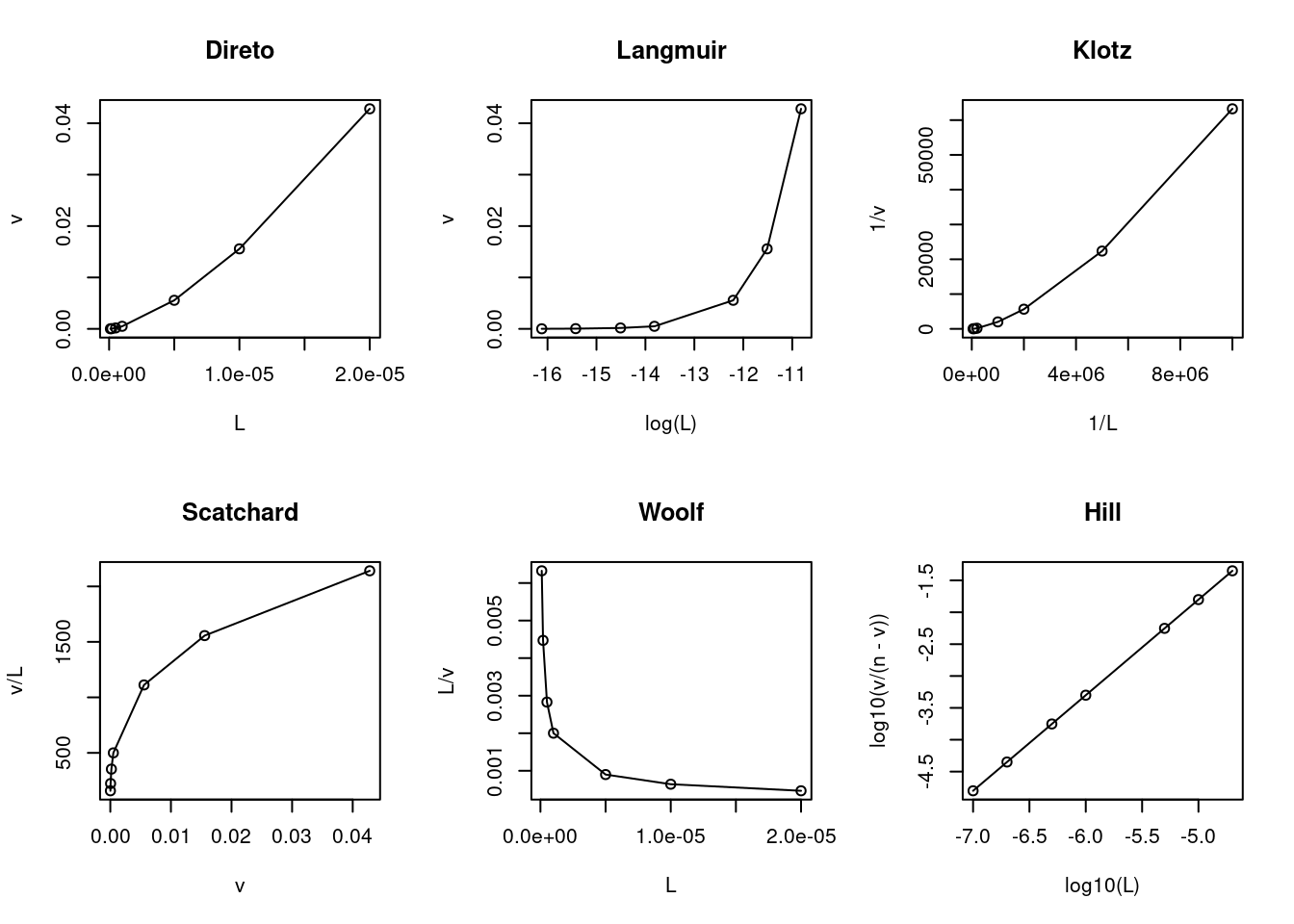
Figura 6.5: Modelo e linearizações para cooperatividade positiva de sítios de ligação.
par(mfrow=c(1,1)) # volta à janela gráfica normalAinda que sujeito à crítica por sua inconsistência estatística (variável dependente em ambos os eixos), a representação de Scatchard tem sido privilegiada ao longo de décadas como diagnóstico de modelos de interação ligante-proteína. Entre suas vantagens, aloca-se a possibilidade de facilmente distinguir-se o modelo de cooperatividade positiva (aclive) do de heterogeneidade de sítios de ligação (declive abrupto) ou de cooperatividade negativa (declive suave).
6.2 Ajuste Não-Linear Em Interação Ligante-Proteína
# Isoterma de Interação Ligante-Proteína
n=1
Kd=10
L=120
i=3
L=seq(0, L, i)
v=(L*n)/(Kd+L)+rnorm(40,0,0.1)## Warning in (L * n)/(Kd + L) + rnorm(40, 0, 0.1): longer object length is not a
## multiple of shorter object lengthplot(L,v)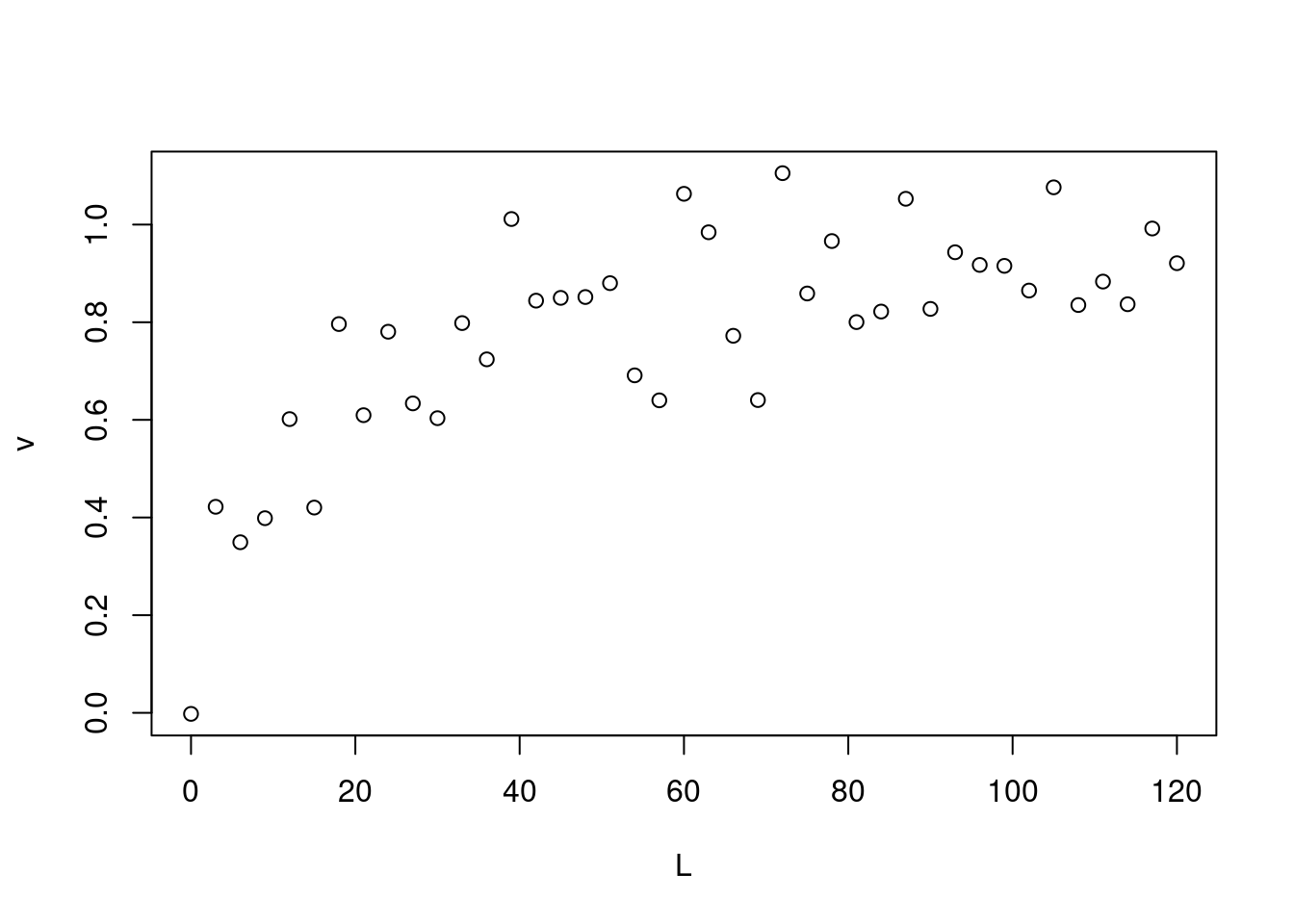
# Simulação de dados de interação bimolecular (1 sítio)
# Simulação de dados
set.seed(20160227) # estabelece semente para geração de números aleatórios
L<-seq(0,50,1)
PL<-((runif(1,10,20)*L)/(runif(1,0,10)+L))+rnorm(51,0,1)
# 1. runif(n,min,max); quando sem atributos, considera-se min=0 e max=1
# 2. rnorm(no. pontos,media,desvio) - erro aleatório de distribuição normal
plot(L,PL, xlab="L", ylab="PL")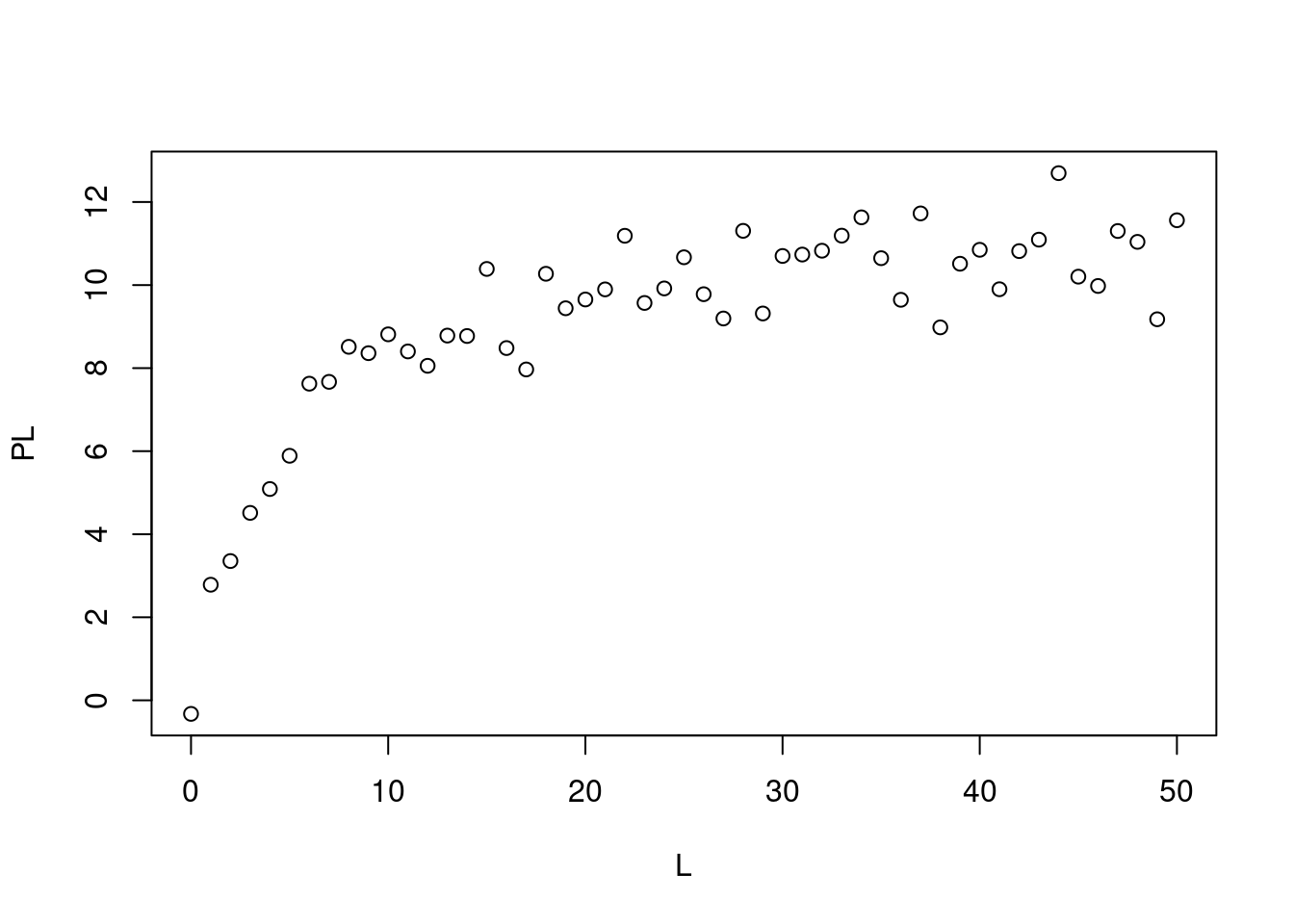
Figura 6.6: Dados simulados para isoterma de interação bimolecular.
#Ajuste não linear
m<-nls(PL~n*L/(Kd+L),start=list(n=1,Kd=1))
#Coef. de correlação
cor(PL,predict(m)) # Coeficiente de correlação de Pearson## [1] 0.9#Gráfico de dados e simulação
plot(L,PL)
lines(L,predict(m),lty=2,col="red",lwd=3)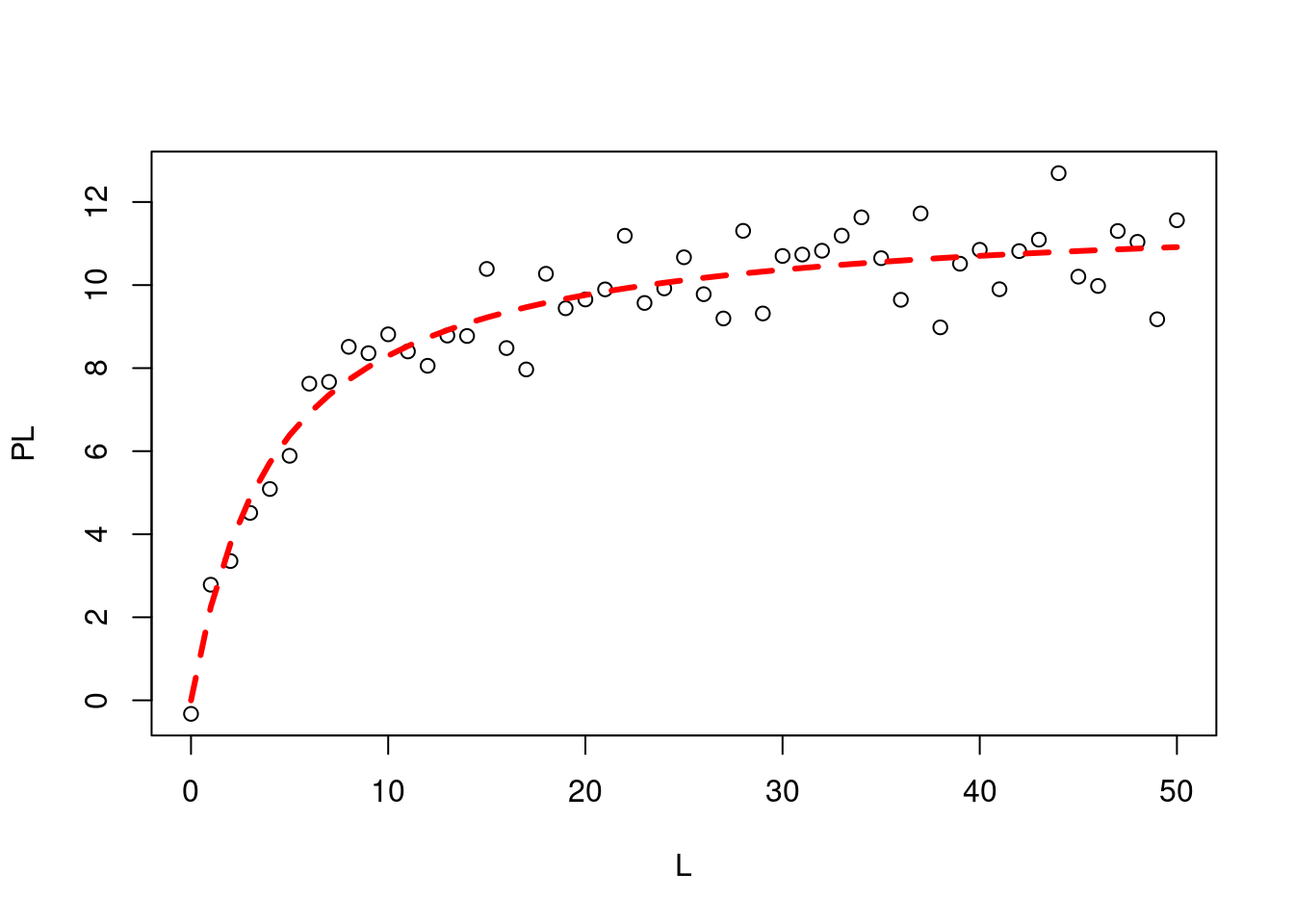
summary(m)##
## Formula: PL ~ n * L/(Kd + L)
##
## Parameters:
## Estimate Std. Error t value Pr(>|t|)
## n 11.848 0.262 45.22 < 2e-16 ***
## Kd 4.278 0.511 8.37 5.3e-11 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.8 on 49 degrees of freedom
##
## Number of iterations to convergence: 6
## Achieved convergence tolerance: 1.55e-066.3 Sistemas Gráficos no R
O sistema Lattice (Sarkar 2008) é baseado no sistema Trellis para representação gráfica de dados multivariados. Sua força está na representação de dados em paineis contendo subgrupos e, embora tenha sintaxe menos intuitiva e por vezes mais elaborada que o pacote Graphics, produz um grafismo superior a esse com poucos cliques de teclado. De modo geral, o Lattice produz o gráfico dentro do próprio algoritmo, de modo diferente aos sistemas Graphics (pode-se acumular linhas sucessivas de modificação do gráfico) ou ggplot2.
Por outro lado a biblioteca ggplot2 é baseada na gramática de gráficos (Wickham 2011), e produz o gráfico utilizando uma única linha de comando que combina camadas sobrepostas, de modo similar à aplicativos de manipulação de imagens (ex: Inkscape, Gimp, Corel Draw, Photoshop). Dessa forma é possível alterar cada ítem do gráfico em suas camadas específicas (tema, coordenadas, facets, estatísticas, geometria, estética, dados). Exemplificando o resultado gráfico da curva de simulação acima de binding para Lattice e ggplot2:
# Simulação de dados
set.seed(20160227) # estabelece semente para geração de números aleatórios
L<-seq(0,50,1)
PL<-((runif(1,10,20)*L)/(runif(1,0,10)+L))+rnorm(51,0,1)
# 1. runif(n,min,max); quando sem atributos, considera-se min=0 e max=1
# 2. rnorm(no. pontos,media,desvio) - erro aleatório de distribuição normal
# Produção do gráfico com sistema Lattice
library(lattice)
xyplot(PL~L)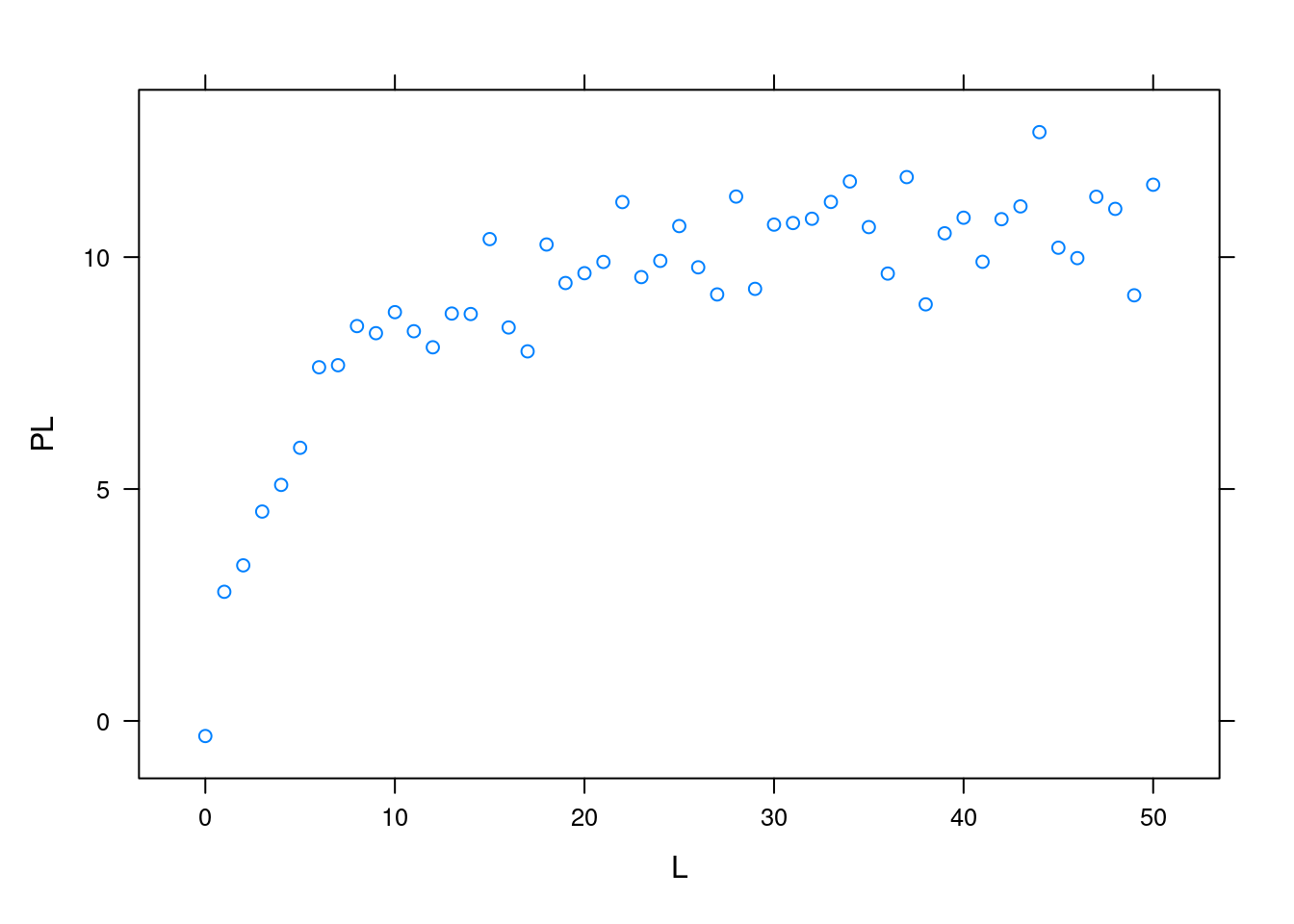
# Produção do gráfico com sistema ggplot2
library(ggplot2)
qplot(L,PL)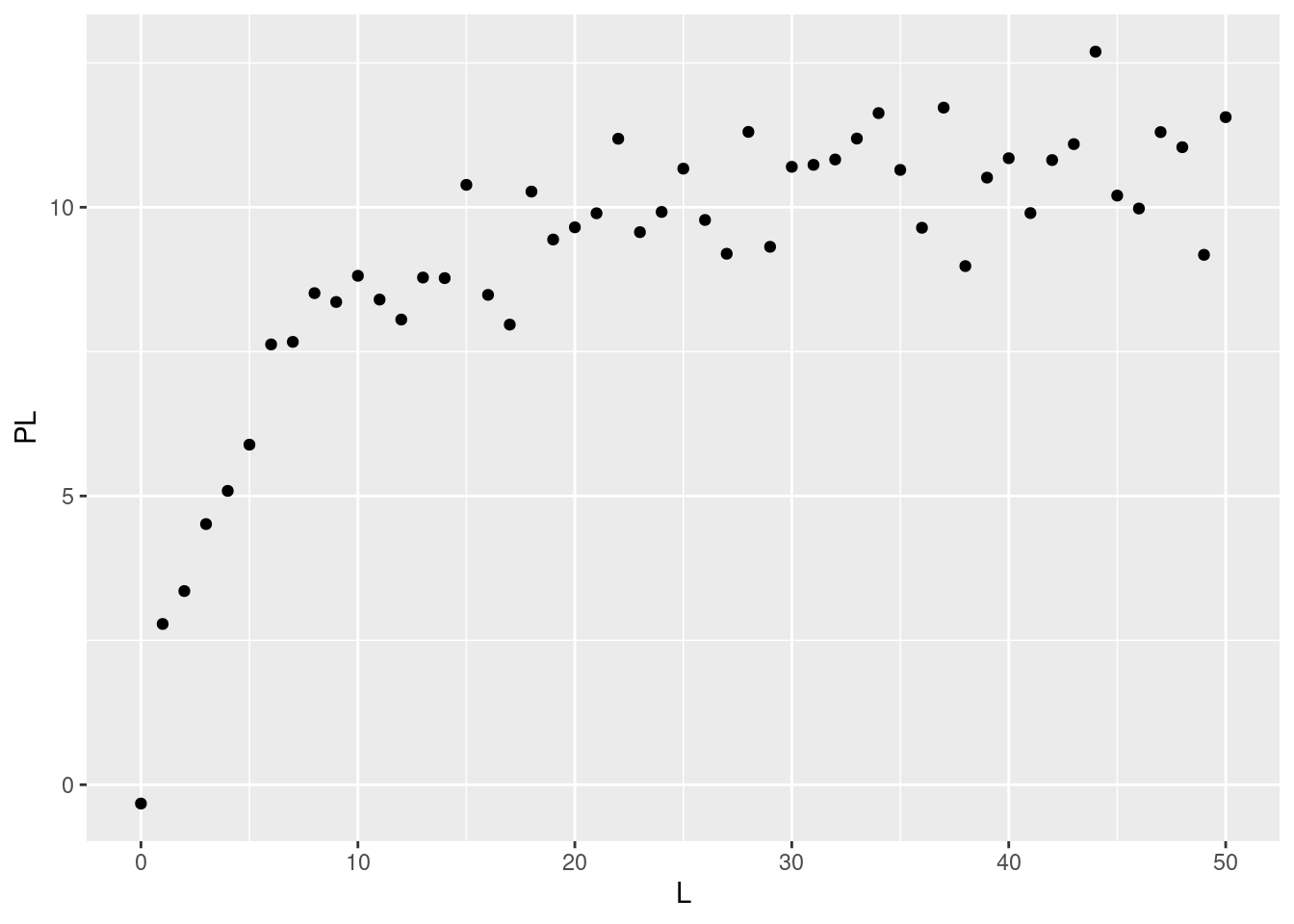
library(datasets)
plot(rate ~ conc, data = Puromycin, las = 1,
xlab = "[S], mM",
ylab = "v (contagem/min/min)",
pch = as.integer(Puromycin$state),
col = as.integer(Puromycin$state),
main = "Ilustração de Ajuste Com Graphics")
## Ajuste da equação de Michaelis-Mentem
fm1 <- nls(rate ~ Vm * conc/(K + conc), data = Puromycin,
subset = state == "treated",
start = c(Vm = 200, K = 0.05))
fm2 <- nls(rate ~ Vm * conc/(K + conc), data = Puromycin,
subset = state == "untreated",
start = c(Vm = 160, K = 0.05))
summary(fm1)##
## Formula: rate ~ Vm * conc/(K + conc)
##
## Parameters:
## Estimate Std. Error t value Pr(>|t|)
## Vm 2.13e+02 6.95e+00 30.61 3.2e-11 ***
## K 6.41e-02 8.28e-03 7.74 1.6e-05 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 11 on 10 degrees of freedom
##
## Number of iterations to convergence: 5
## Achieved convergence tolerance: 8.82e-06summary(fm2)##
## Formula: rate ~ Vm * conc/(K + conc)
##
## Parameters:
## Estimate Std. Error t value Pr(>|t|)
## Vm 1.60e+02 6.48e+00 24.73 1.4e-09 ***
## K 4.77e-02 7.78e-03 6.13 0.00017 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 10 on 9 degrees of freedom
##
## Number of iterations to convergence: 5
## Achieved convergence tolerance: 4.45e-06## Adição de linhas de ajuste ao plot
conc <- seq(0, 1.2, length.out = 101)
lines(conc, predict(fm1, list(conc = conc)), lty = 1, col = 1)
lines(conc, predict(fm2, list(conc = conc)), lty = 2, col = 2)
legend(0.8, 120, levels(Puromycin$state),
col = 1:2, lty = 1:2, pch = 1:2)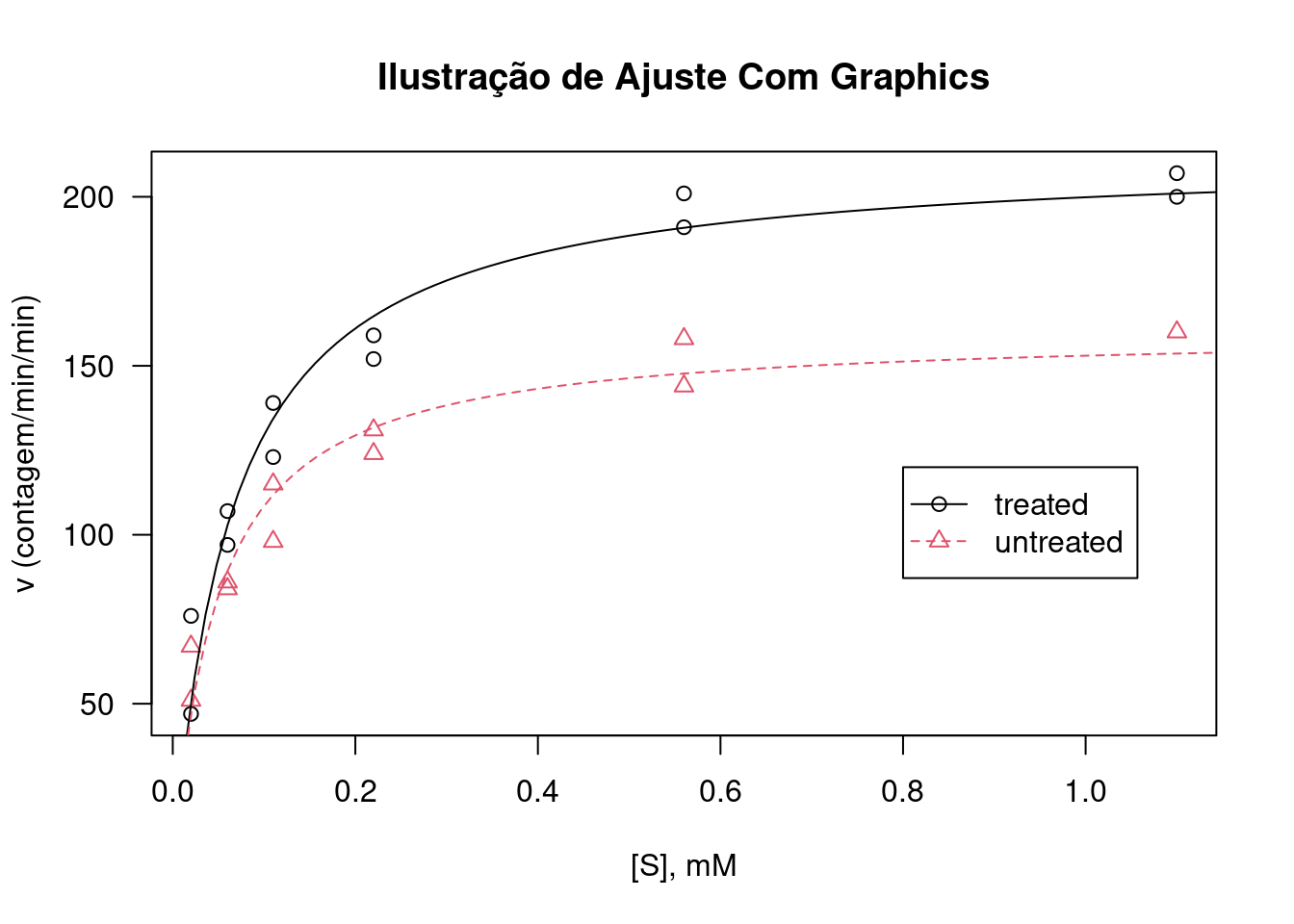
Figura 6.7: Plotagem e análise com graphics.
library(datasets)
p <- ggplot(data=Puromycin, aes(conc, rate, color = state)) +
geom_point() +
geom_smooth(
method = "nls",
formula = y ~ Vm*x/(Km+x),
method.args = list(start = list(Vm = 200, Km = 0.1)),
se = FALSE
) # expressão que define o plot
p # variável que apresenta o plot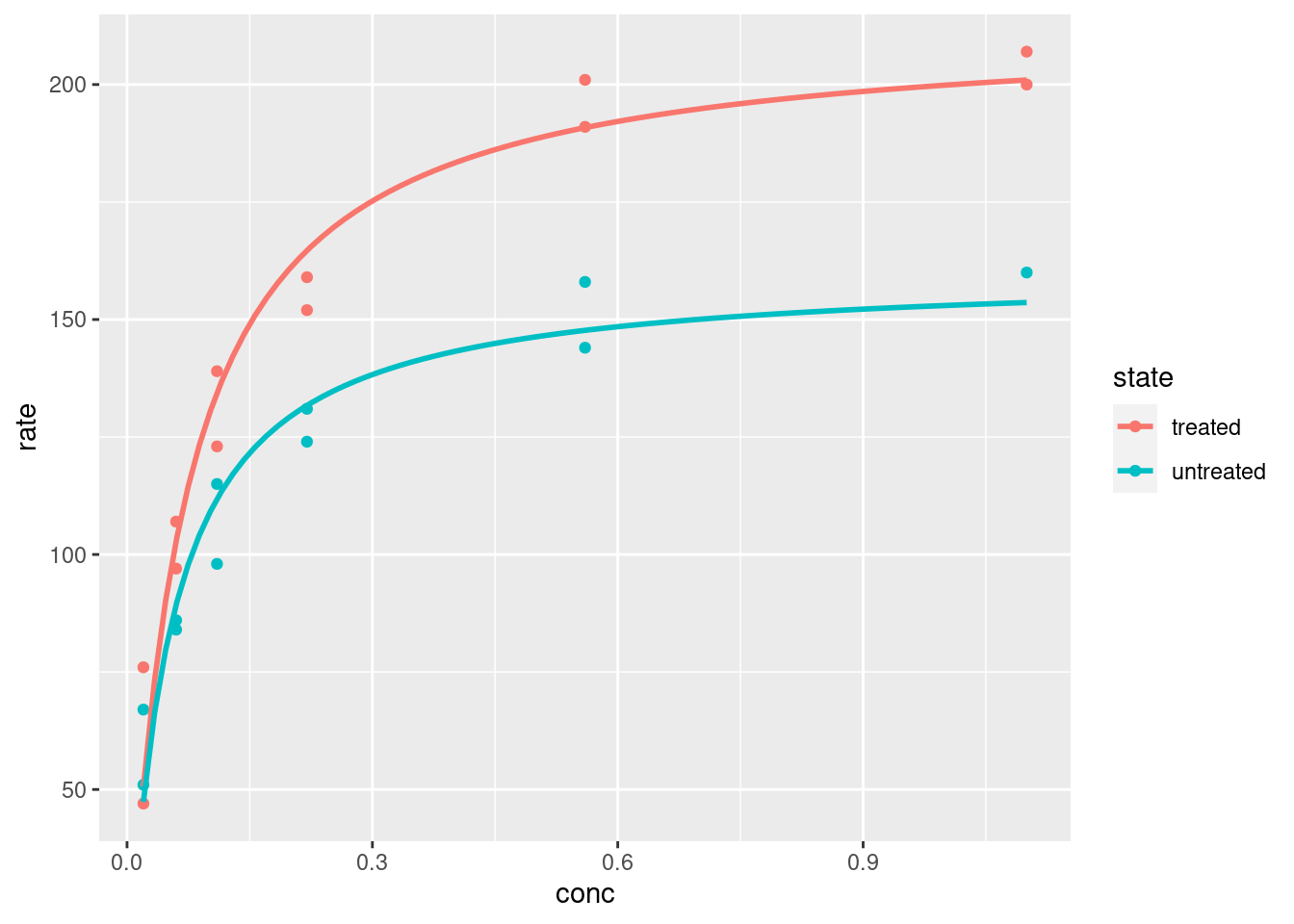
Figura 6.8: Plotagem e análise com ggplot2.
p + facet_grid(rows = vars(state))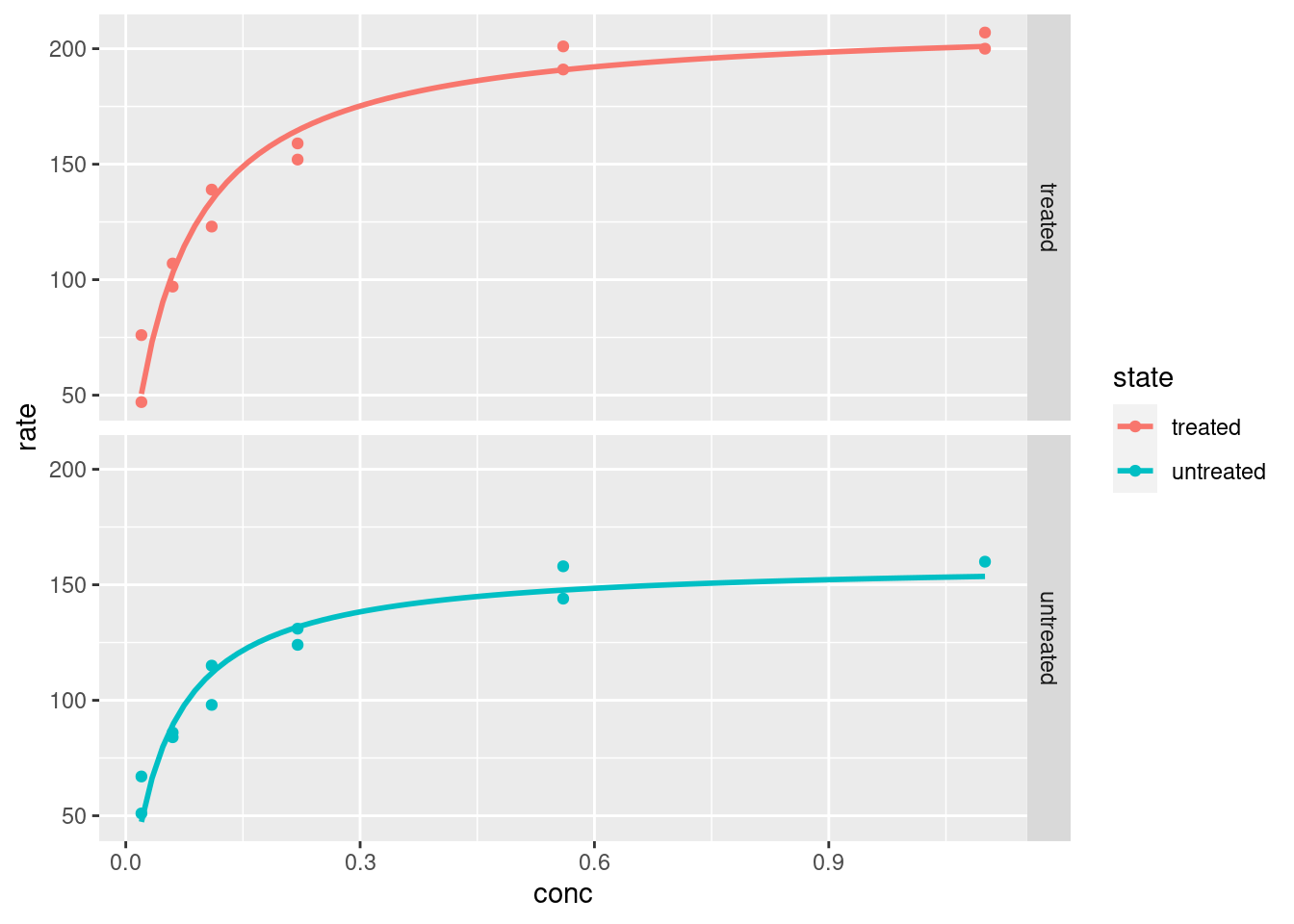
Figura 6.9: Plotagem e análise com ggplot2 - paineis (faceting).
library(lattice)
xyplot(rate ~ conc, data = Puromycin, groups=state)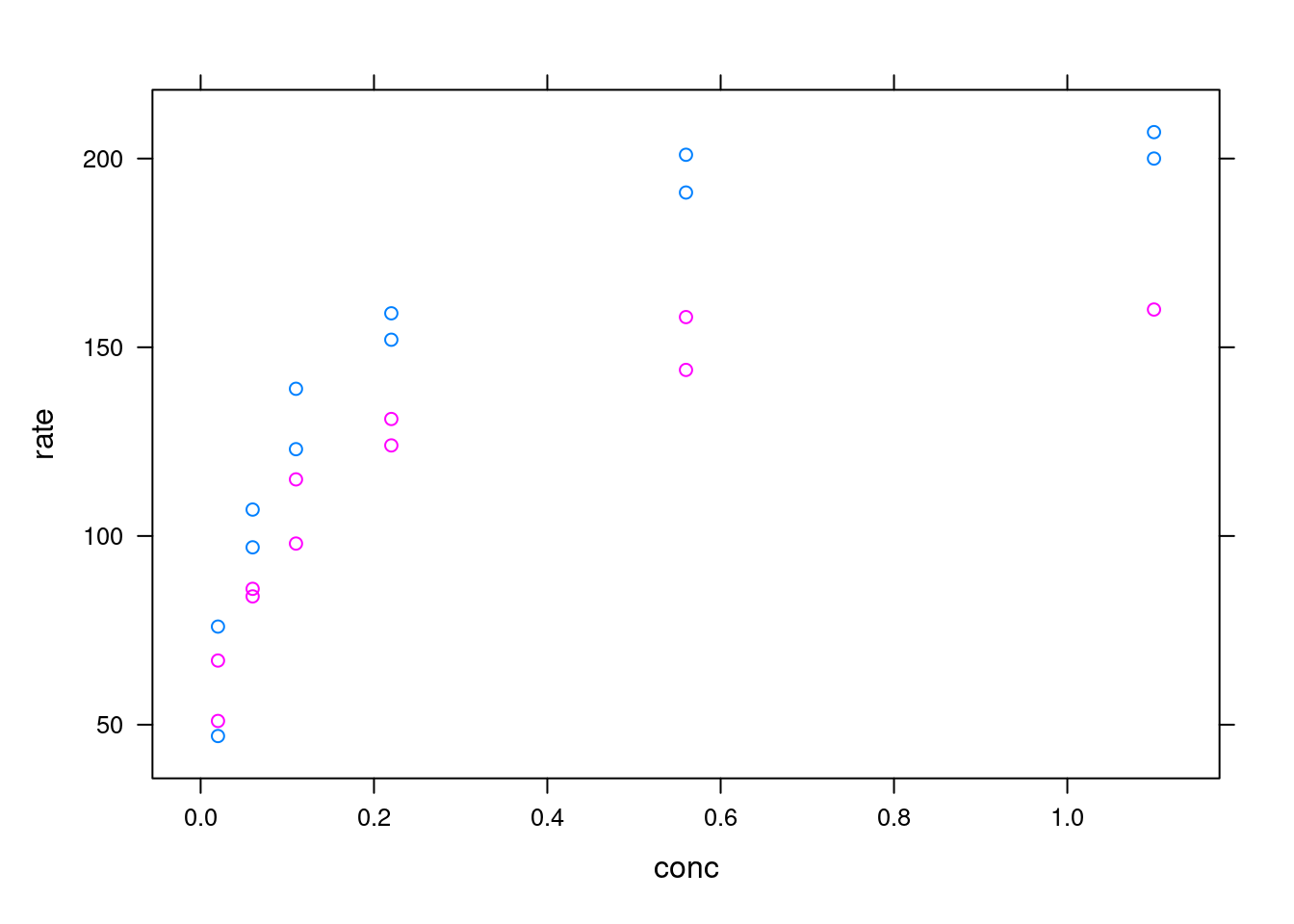
library(nlme)
n1<-nlsList(rate ~ Vmax*conc/(Km+conc) | state, data=Puromycin, start=list(Vmax=200, Km=0.1))
summary(n1)
xyplot(rate ~ conc,groups = state, data =
Puromycin) +
layer(panel.curve(Vmax[1]*x/(Km[1]+x),col=1),
data = as.list(coef(n1))) +
layer(panel.curve(Vmax[2]*x/(Km[2]+x),col=2),
data = as.list(coef(n1)))library(nlme) # pacote quer permite regressão não linear com subgrupos##
## Attaching package: 'nlme'## The following object is masked from 'package:dplyr':
##
## collapse## The following object is masked from 'package:seqinr':
##
## glsnonlinLatt<-nlsList(rate~Vmax*conc/(Km+conc)| state, start=list(Vmax=200, Km=0.1), data=Puromycin)
summary(nonlinLatt)## Call:
## Model: rate ~ Vmax * conc/(Km + conc) | state
## Data: Puromycin
##
## Coefficients:
## Vmax
## Estimate Std. Error t value Pr(>|t|)
## treated 213 7 32 3e-11
## untreated 160 7 23 1e-09
## Km
## Estimate Std. Error t value Pr(>|t|)
## treated 0.06 0.008 8 2e-05
## untreated 0.05 0.008 6 2e-04
##
## Residual standard error: 10 on 19 degrees of freedomxyplot(rate + fitted(nonlinLatt)~conc| state, data=Puromycin,
type=c("p","l"),distribute.type=TRUE,col.line="red",
ylab="rate")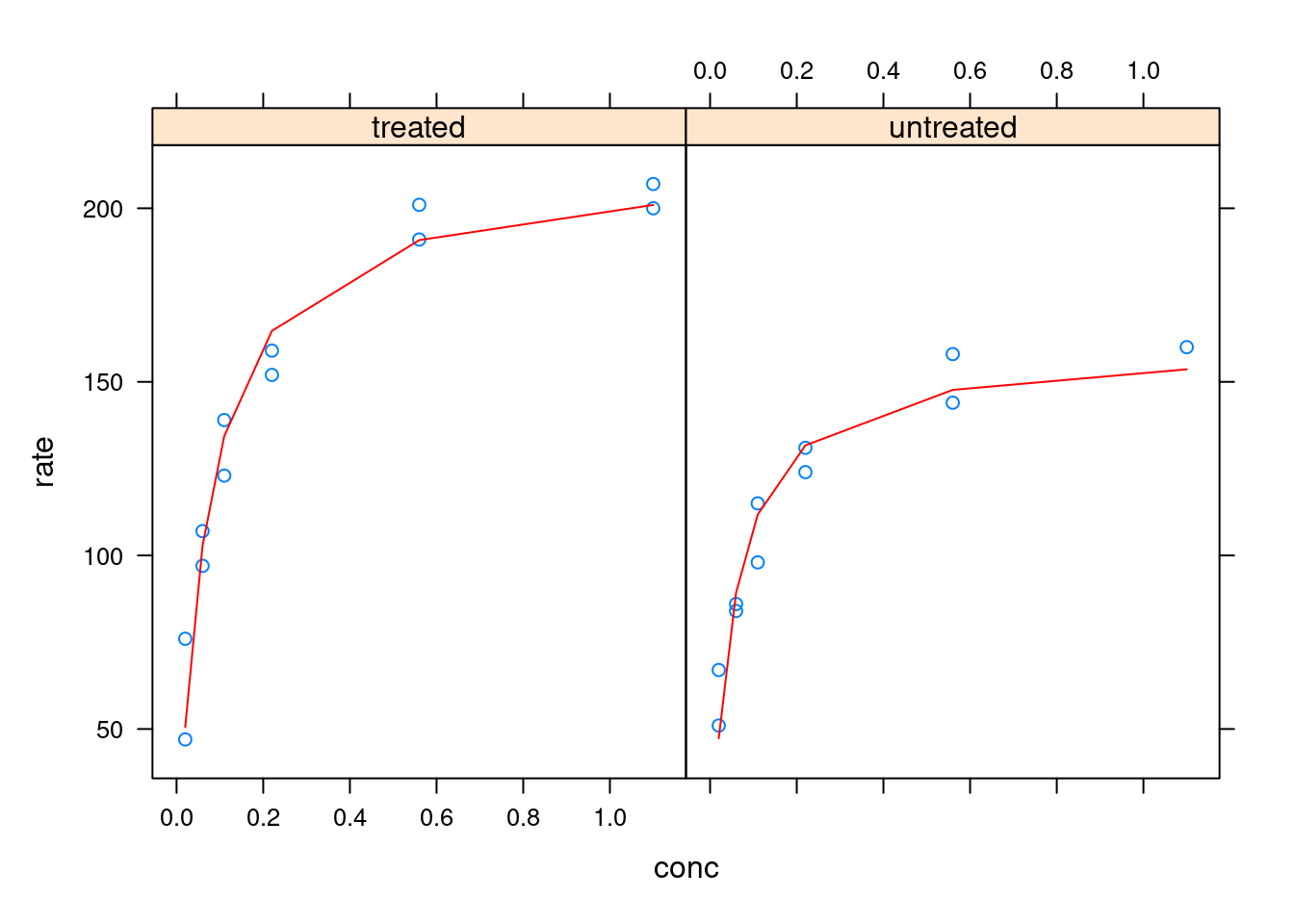
Figura 6.10: Plotagem e análise com Lattice - ajuste externo e paineis.
De certa forma, ainda que o Lattice exija uma curva de aprendizado menos intutiva, ele permite que se utilize os resultados estatísticos obtidos anteriormente para inclusão no algoritmo de plotagem. Isso é vantajoso quando se deseja outros algoritmos estatísticos para ajuste, como acima, ou mesmo sua flexibilização, além do ‘nls’ incluido em ggplot2. Não obstante, o Lattice também permite que se inclua a linha de ajuste dentro do próprio algoritmo, como abaixo:
xyplot(rate ~ conc | state, data = Puromycin,
panel = function(x, y, ...) {
panel.xyplot(x, y, ...)
n3<-nls(y ~ Vmax*x/(Km+x), data=Puromycin, start=list(Vmax=200, Km=0.1))
panel.lines(seq(0.02,1.1,0.02), predict(n3,newdata=data.frame(x=seq(0.02,1.1,0.02))), col.line = 2)
},
xlab = "conc", ylab = "rate")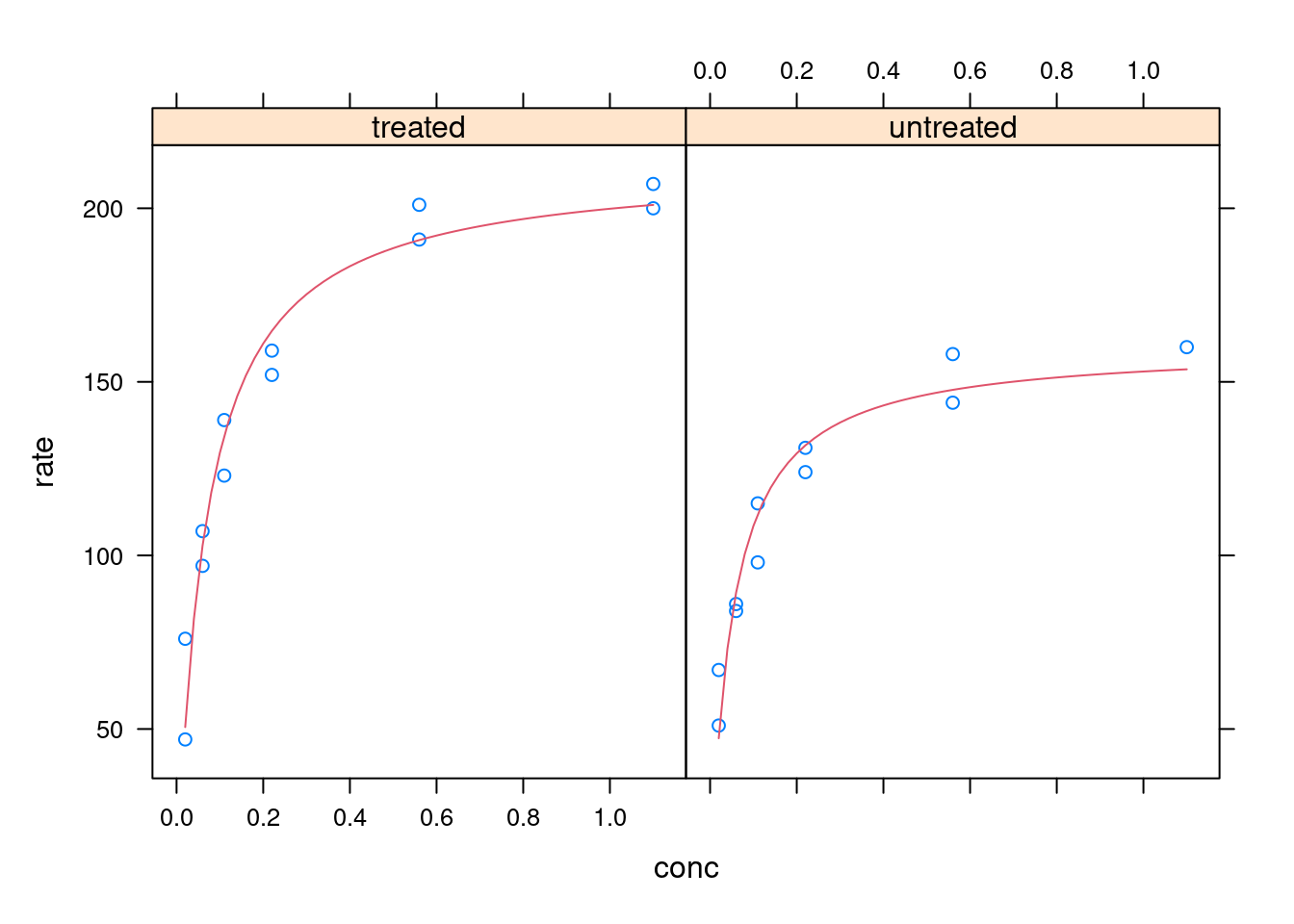
Figura 6.11: Plotagem e análise com Lattice - ajuste interno e paineis.
6.4 Solução Numérica Para o Equilíbrio de Complexos Ligante-Proteína
Usualmente o tratamento dado para a solução numérica envolve encontrar as raízes de uma equação ou sistema de equações, ou seja:
R por diversas maneiras, uma das quais pela função de minimização rootSolve:#Cálculo de L, P, e PL em interação biomolecular para 1 conjunto de sítios de mesma afinidade
library(rootSolve)
Pt =1; Lt=10; Kd=4
# Modelo
model = function(x) c(x[1]+(x[1]*x[2])/Kd-Pt,x[2]+(x[1]*x[2])/Kd-Lt,Pt-x[1]-x[3])
# o modelo acima deve conter uma lista de equações cuja igualdade é zero, ou seja, f(x)=0
(ss=multiroot(model,c(1,1,1))) # comando de execução do rootSolve (sementes pro algoritmo)## $root
## [1] 0.3 9.3 0.7
##
## $f.root
## [1] 5e-08 5e-08 5e-12
##
## $iter
## [1] 5
##
## $estim.precis
## [1] 4e-08optim do R (limites de busca da solução, emprego de vetores, por ex). Para isso será exemplificado a mesma situação acima, embora apresentando uma variação do formalismo que relaciona P, L e PL :optim do R:#Cálculo de L, P, PL em interação para 1 sítio
model2=function(x,Pt,Lt,K){L=x[1];P=x[2];PL=x[3];(Pt-P-PL)^2+(Lt-L-PL)^2+(P*L-Kd*PL)^2} # declaração da função
Pt=1;Lt=10;Kd=4 # parâmetros da função
sol2num = optim(c(0.5,1,.5),model2,method="L-BFGS-B",lower=c(0,0,0),upper=c(Lt,Pt,Pt),Pt=Pt,Lt=Lt) # método BFGS permite bounds (lower, upper)
sol2num$par # LF, PF, PL calculados## [1] 9.3 0.3 0.7#Declaração da função
bind1=function(x,Pt,Lt,Kd) {
L=x[1];P=x[2];PL=x[3];
(Pt-P-PL)^2+(Lt-L-PL)^2+(P*L-Kd*PL)^2
}
#Parâmetros da função
Pt = 1; Lt = c(5,10,20); Kd = 4
#Minimização (parâmetros para que a função acima dê zero)
y = function(i) optim(c(1, 1, 1), bind1, method =
"L-BFGS-B", lower = c(0,0,0), upper = c(Lt[i], Pt,
Pt), Lt=Lt[i], Pt=Pt, Kd=Kd)
# Resultados em matriz
ypar = function(i) y(i)$par
yp = matrix(nrow=length(Lt), ncol=2+length(Kd),
byrow=T)
for (i in 1:length(Lt)) yp[i,]=y(i)$par
colnames(yp) = c("L","P","PL")
rownames(yp) = c("5","10","20")
yp## L P PL
## 5 4 0.5 0.5
## 10 9 0.3 0.7
## 20 19 0.2 0.86.5 Cinética de Interação Ligante-Proteína e Solução Numérica
R permite solução de mesma natureza para a cinética da formação dos complexos, ou seja, os teores de P, L e PL observados no tempo. Nesse caso pode-se desenvolver outras relações a partir da Eq. (6.1). Tomando-se por base que no equilíbrio as taxas cinéticas de k e k se igualam (steady-state), pode-se relacionar algumas equações diferenciais para a associação, bem como para a dissociação dos complexos:R que permita a solução de um sistema de equações diferenciais. Entre as muitas soluções (odeintr, pracma, rODE), o emprego da biblioteca deSolve, que utiliza uma função para integração do sistema por algoritmo de Runge-Kutta de 4a. ordem:# Cinética de interação ligante-protéina para 1 conjunto de sítios
library(deSolve)
# Condições experimentais
tempo = seq(0, 100) # intervalo de tempo
parms = c(kon=0.02, koff=0.001) # parâmetros do estado estacionário da interação (uM^-1*s^-1 e s^-1, respectivamente)
val.inic = c(L=0.8, P=1, PL=0) # valores iniciais, uM
# Integração do sistema por Runge-Kutta de 4a. ordem
solNumKin = function(t, x, parms) {
# definição da lista de parâmetros
L = x[1] # ligante
P = x[2] # proteína
PL = x[3] # complexo
with(as.list(parms),{
# definição da lista de equações diferenciais
dL = -kon*L*P + koff*PL
dP = -kon*L*P + koff*PL
dPL = kon*L*P-koff*PL
res<-c(dL, dP, dPL)
list(res)
})
}
sol.rk4 = as.data.frame(rk4(val.inic, tempo, solNumKin,
parms)) # rotina para Runge-Kutta 4a. ordem
# Gráfico
plot (sol.rk4$time, sol.rk4$L, type="l", xlab="tempo",ylab="[composto], uM")
legend("topright",c("L","P","PL"),text.col=c("black","red","blue"),bty="n",lty=c(1,2,3))
lines (sol.rk4$time, sol.rk4$P, type="l", lty=2,col=2,lwd=1.5)
lines (sol.rk4$time, sol.rk4$PL, type="l", lty=3,col=3,lwd=1.5)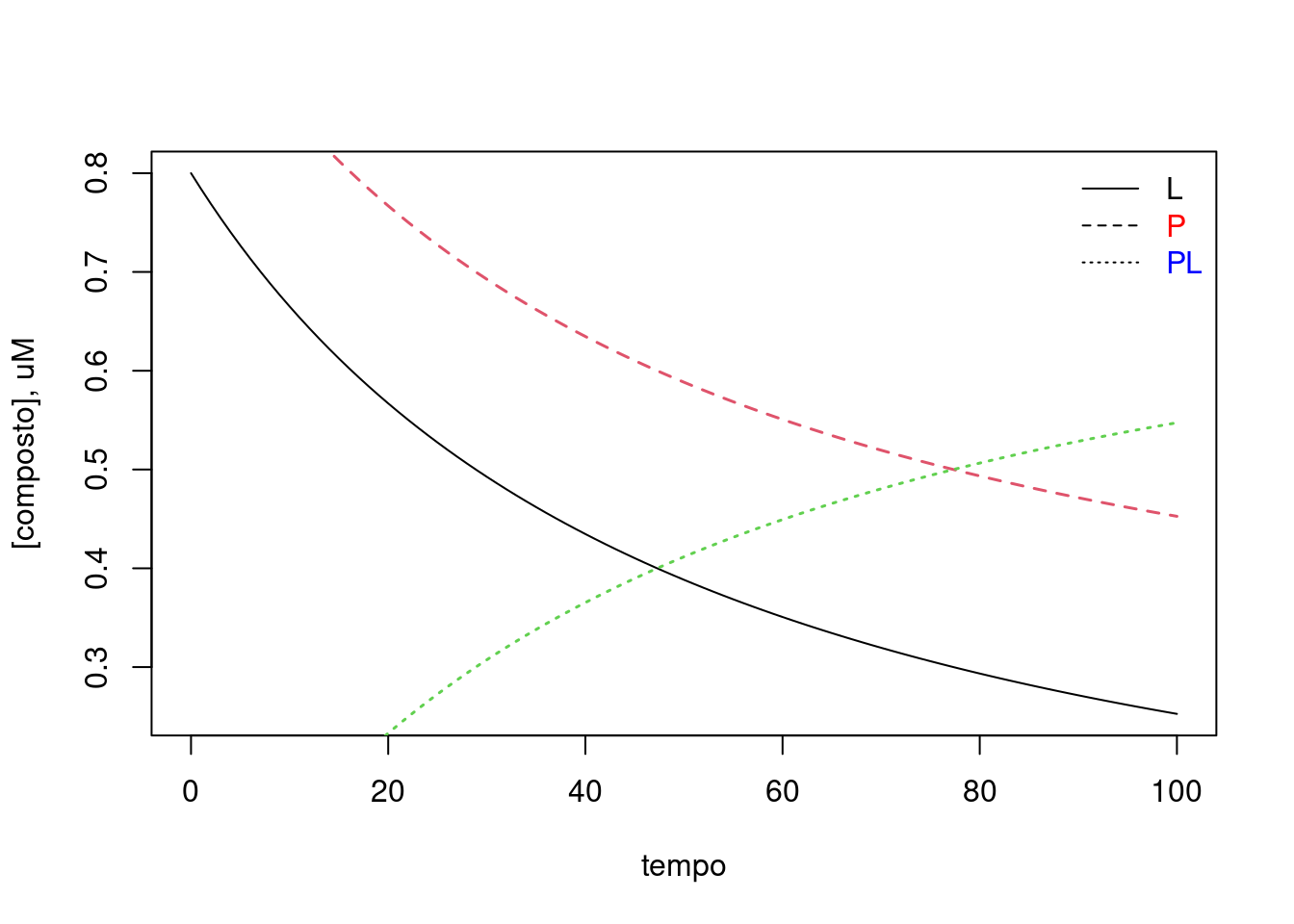
Figura 6.12: Teores de ligante e proteína livres (L e P), bem como do complexo PL apresentados ao longo do tempo de acordo com o método Runge-Kutta de 4a. ordem para solução de equações diferenciais.