Capítulo 3 Aminoácidos & Peptídios
3.1 Ponto isoelétrico & aminoácidos
Como todos os 20 aminoácidos que participam da estrutura proteica possuem grupos ionizáveis, tanto em seu esqueleto carbônico como em sua cadeia lateral, é possível prever o ponto isoiônico de um aminoácido em função dos valores de pKa apresentados nesses grupos ionizáveis. O pI também é denominado comumente por ponto isoelétrico, embora essa definição encerre em si uma abrangência teórica mais complexa.
Exemplificando, o ácido glutâmico (Glu, E) apresenta um carboxilato ionizável em sua cadeia lateral, além dos grupos amina (-H\(_2\)N) e carboxilato do esqueleto carbônico (Figura 3.1):
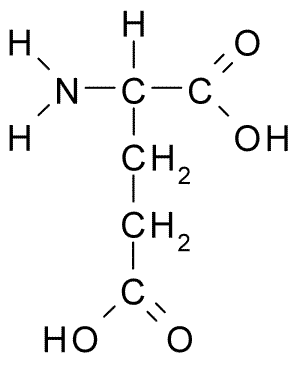
Figura 3.1: Fórmula estrutural planar para o ácido glutâmico (Wikimedia).
, com pKi como o enésimo valor de pKa. Dessa forma pode-se determinar programaticamente a curva de titulação do ácido glutâmico em função de sua carga, e não da fração ácida. Nessa linha, qb representa a forma do composto em base, o que para Glu apresentará os valores de -1 para os dois carboxilatos, e de 0 para o grupo amina, sendo necessário compor um vetor adicional para qb.
#Titulação de Glu
qNet = function(pH, qB, pKa) {
x=0
for(i in 1:length(qB)) {
x = x+qB[i] + 1/(1 + 10^(pH - pKa[i]))}
return(x)
}
qB=c(-1,0,-1)
pKa=c(2.2, 9.7, 4.3)
curve(qNet(x,qB,pKa),1,12, xlab="pH", ylab="qNet")
abline(0,0,lty="dotted")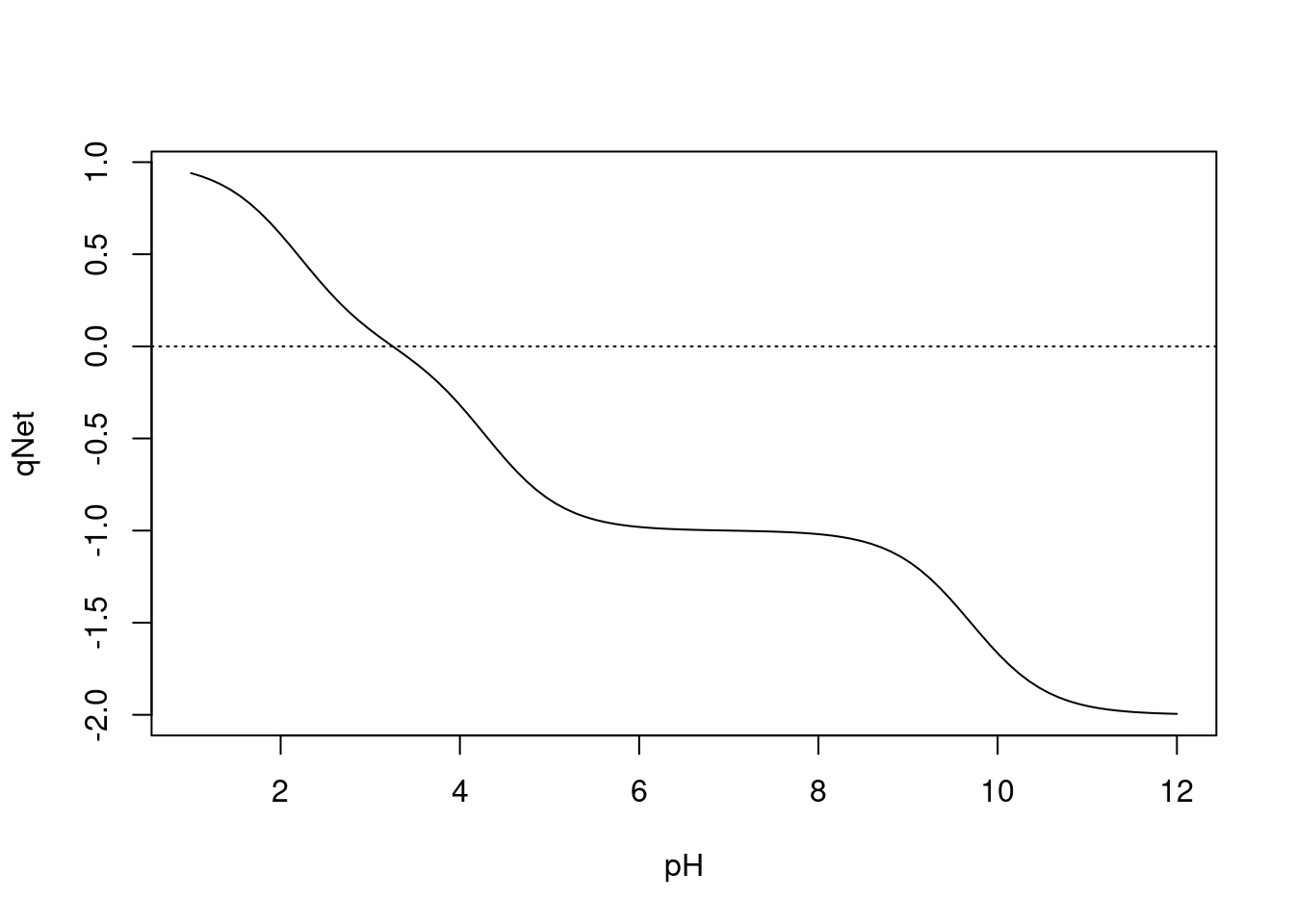
Figura 3.2: Curva de titulação para o ácido glutâmico. A curva intercepta a linha pontilhada no ponto isoiônico do Glu.
locator() visto anteriormente. Mas também é possível acessar esse valor automaticamente, aplicando um comando que encontre a raíz dessa função, ou seja, o valor de pH que corresponda a um valor nulo para qnet. Para isto, exemplifica-se o uso de uniroot, no qual define-se a função matemática pretendida, bem como os limites inferior e superior para a busca pelo algoritmo, como segue: #Cálculo de pI
f=function(pH) {qNet(pH,qB,pKa)}
str(uniroot(f,c(2,5)))## List of 5
## $ root : num 3.25
## $ f.root : num -4.8e-06
## $ iter : int 4
## $ init.it : int NA
## $ estim.prec: num 6.1e-05root), em 4 iterações, com uma estimativa de precisão de 6,1x10^{-5}, e erro associado de -4,8x10^{-6}.Essa forma de se obter um valor empregando-se o cálculo numérico é por vezes denominada solução numérica. Por outro lado, pode-se obter o valor de pI para o Glu por um procedimento mais simples, normalmente encontrado nos livros-texto sobre o assunto, e que assume a forma abaixo:
3.2 Ponto isoiônico & biopolímeros
# Titulação de Lisozima e Determinação de pI
#Define função para qNet
qNet = function(pH, qB, pKa, n) {
x=0
for(i in 1:length(qB)) {
x = x+n[i]*qB[i] + n[i]/(1 + 10^(pH - pKa[i]))}
return(x)
}
# Define pKas de aCOOH, aNH3 e as 7 cadeias laterais de AA
pKa=c(2.2, 9.6, 3.9, 4.1, 6.0, 8.5, 10.1, 10.8, 12.5)
# Define qB, as cargas de cada aminoácido na forma básica
qB =c(-1, 0, -1, -1, 0, -1, -1, 0, 0)
ionizavel=c("aCOOH","aNH3","Asp","Glu","His","Cys","Tyr",
"Lys","Arg")
n=c(1,1,7,3,1,8,6,5,14) # Lista para quantidades de resíduos ionizáveis na lisozima (cada elemento representa a quantidade de aCOOH, aNH3, e determinado AA na enzima)
# Cálculo de pI
f=function(pH) {qNet(pH,qB,pKa,n)}
str(uniroot(f,c(1,13))) # estimativa de pI entre 10 e 12## List of 5
## $ root : num 9.46
## $ f.root : num 3.3e-07
## $ iter : int 7
## $ init.it : int NA
## $ estim.prec: num 6.1e-05# Gráfico de titulação
curve(qNet(x,qB,pKa,n),1,12, xlab="pH", ylab="qNet")
abline(0,0,lty=3)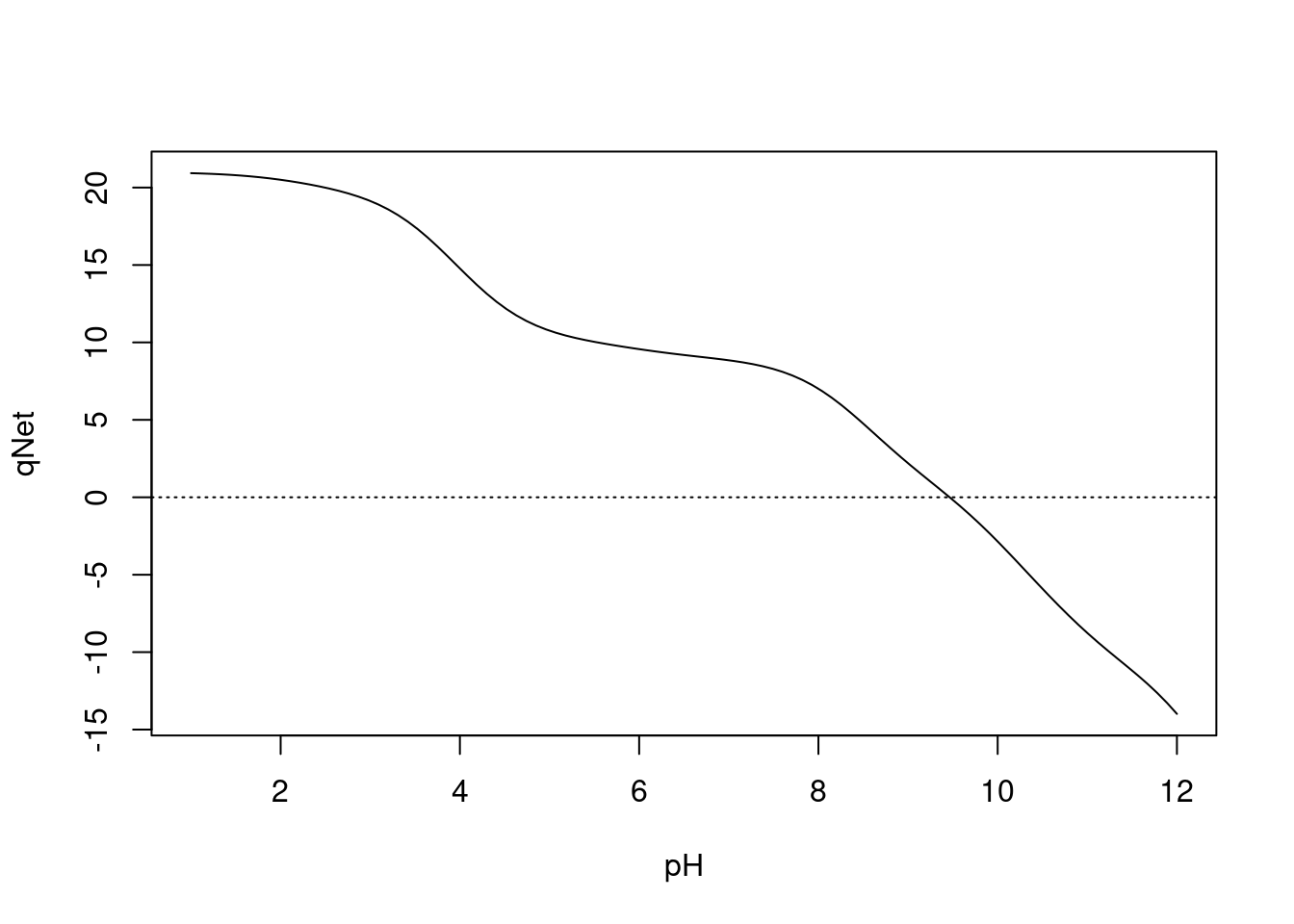
Figura 3.3: Curva de titulação para lisozima.
3.3 Ponto isoiônico & bibliotecas do R
packages), não sendo diferente para determinção de propriedades de biopolímeros, tais como pI.Entre as bibliotecas existentes para propriedades físico-químicas de proteínas e ácidos nucleicos exemplifica-se o pacote
seqinr, Biological Sequences Retrieval and Analysis 2, de análise e visualização exploratória de biopolímeros. Para uso desse pacote, contudo, faz-se necessário a obtenção da sequência primária da proteína, representada em código de uma letra. Pode-se obter a sequência primária da lisozima pelo sítio do National Center for Biotechnology Information, NCBI 3. Um truque rápido envolve:digitar o nome da proteína;
selecionar entre as opções resultantes;
clicar em FASTA para obter a sequência primária de 1 letra.
copiar a sequência da proteína apresentada para o
seqinr.
seqinr esteja instalada, e que a sequência tenha sido obtida para a lisozima (busca por CAA32175 ou lysozyme [Homo sapiens]), pode-se encontrar o valor do pI para a mesma pelo código que segue:library(seqinr)
lisozima<-s2c("KVFERCELARTLKRLGMDGYRGISLANWMCLAKWESGYNTRATNYNAGDR STDYGIFQINSRYWCNDGKTPGAVNACHLSCSALLQDNIADAVACAKRVV RDPQGIRAWVAWRNRCQNRDVRQYVQGCGV") # converte sequência de string em vetor de caracteres
computePI(lisozima)## [1] 9.2778seqinr. Exemplificando essa variação, o próprio seqinr apresenta valores de pKa diferentes, em função da base de dados buscada. Para verificar isso, digite o comando abaixo e visualize e variável pK resultante.library(seqinr)
data(pK)seqinr do R.library(knitr)
knitr::kable(pK, "pipe", caption = "Tabela de valores de pKa para aminoácidos a partir de diversas fontes, extraída do pacote seqinr.")| Bjellqvist | EMBOSS | Murray | Sillero | Solomon | Stryer | |
|---|---|---|---|---|---|---|
| C | 9.00 | 8.5 | 8.33 | 9.0 | 8.3 | 8.5 |
| D | 4.05 | 3.9 | 3.68 | 4.0 | 3.9 | 4.4 |
| E | 4.45 | 4.1 | 4.25 | 4.5 | 4.3 | 4.4 |
| H | 5.98 | 6.5 | 6.00 | 6.4 | 6.0 | 6.5 |
| K | 10.00 | 10.8 | 11.50 | 10.4 | 10.5 | 10.0 |
| R | 12.00 | 12.5 | 11.50 | 12.0 | 12.5 | 12.0 |
| Y | 10.00 | 10.1 | 10.07 | 10.0 | 10.1 | 10.0 |
Pacote seqinr: https://cran.r-project.org/web/packages/seqinr/index.html↩︎
pacote Peptides. https://cran.r-project.org/web/packages/Peptides/index.html↩︎