Quando se menciona interação entre biomoléculas, normalmente se faz referência à processos adsortivos envolvendo um biopolímero (proteína, ácido nucleico, glicano), e um ligante de baixo peso molecular, embora o formalismo também se aplique com alguma restrição a interações entre biopolímeros, e mesmo células inteiras.
O formalismo mais comum para interação biomolecular é o que envolve a formação de complexo adsortivo entre uma proteína e um ligante (ligand binding), exemplificado para íons (Ca\(^{2+}\), Mg\(^{2+}\), etc), fármacos e candidatos, produtos naturais, e antígenos, dentre vários.
Perguntas simples acerca da interação ligante-proteína podem elucidar diversas características da formação de tais complexos, como:
Quanto de proteína/ligante estão presentes ?
Quanto do complexo é formado ?
Quão rápido o complexo associa/dissocia ?
Quais os mecanismos envolvidos ?
De modo geral, pode-se representar a interação ligante-proteína como segue:
Onde P representa o teor de proteína livre, L o ligante livre, e PL o complexo formado. As taxas de reação são definidas para a formação (k\(_{on}\); M\(^{-1}\)s\(^{-1}\)) e dissociação (k\(_{off}\); s\(^{-1}\)) do complexo.
Dessa forma deduz-se a equação para a isoterma de interação do ligante com a proteína como segue:
\[
Kd=\frac{[P]*[L]}{[P]+[L]}
\tag{2}\]
Onde Kd representa a constante de equilíbrio de dissociação para o complexo PL formado, tal como condicionado ao equilíbrio de formação/dissociação do complexo (v\(_{assoc}\) = v\(_{dissoc}\)), e também definido como:
\[
Kd=\frac{k_{off}}{k_{on}}
\tag{3}\]
A partir da Equação 2 pode-se facilmente deduzir a expressão final para a interação de um ligante a um conjunto de sítios de mesma afinidade na proteína:
\[
\nu=\frac{n * [L]}{Kd + [L]}
\tag{4}\]
Modelos de Interação e Representações Lineares
Observe que a Equação 4 praticamente repete o formalismo já visto com a formação do complexo ativado de enzima-substrato do capítulo de Enzimas, bem como sua representação resultante como uma hipérbole quadrática. De fato, ocorre essencialmente a substituição do parâmetro cinético v da reação pelo parâmetro termodinâmico \(\nu\) (“nu”, do Grego) para a isoterma de ligação. As demais quantidades envolvidas mantém-se análogas (P no lugar de E; L no lugar de S;Kd no lugar de Km; e n no lugar de Vmax).
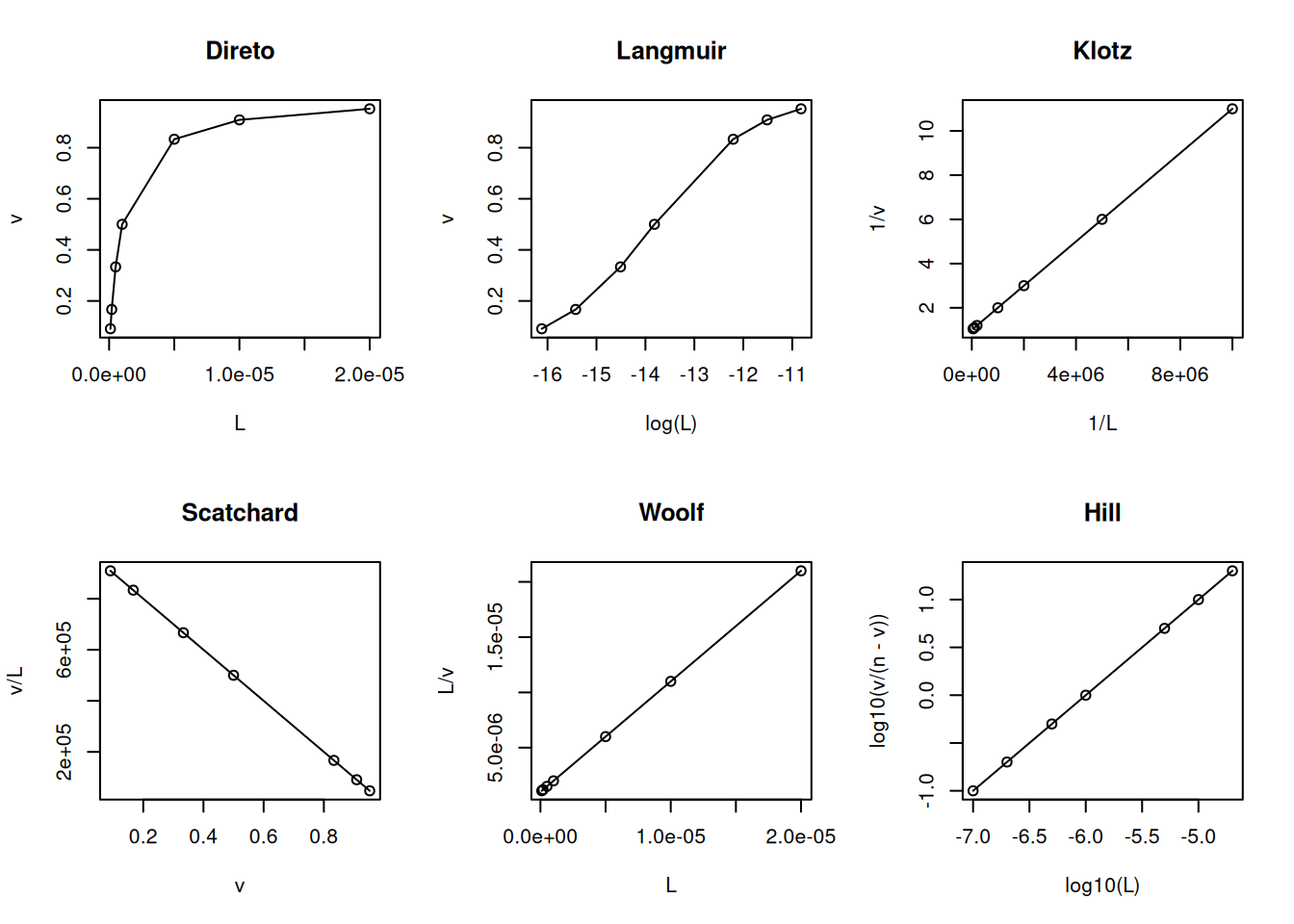
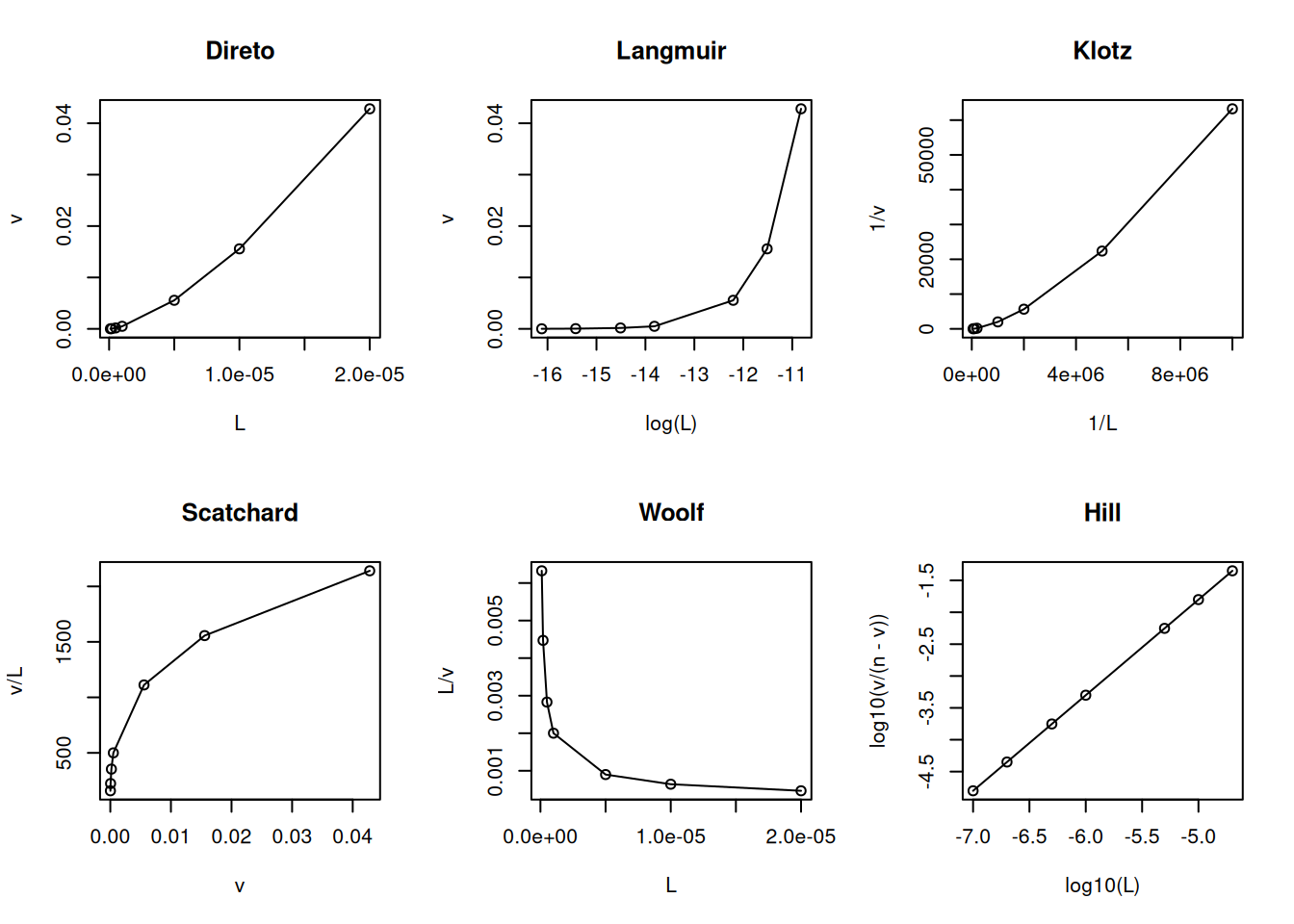
Mantida essa similaridade com o formalismo da equação de Michaelis-Menten, da mesma maneira decorrem as linearizações para a Equação 4), bem como ajustes não lineares à mesma, na busca de uma solução analítica para os parâmetros termodinâmicos Kd e n. Exemplificando um trecho de código para as linearizações mais comuns no tratamento de dados de interação ligante-proteína:
# Linearizações em interação bimolecularL <-c(0.1, 0.2, 0.5, 1, 5, 10, 20) *1e-6Kd <-1e-6n <-1v <- n * L / (Kd + L)par(mfrow =c(2, 3)) # estabelece área de plot pra 6 gráficosplot(L, v, type ="o", main ="Direto")plot(log(L), v, type ="o", main ="Langmuir")plot(1/ L, 1/ v, type ="o", main ="Klotz")plot(v, v / L, type ="o", main ="Scatchard")plot(L, L / v, type ="o", main ="Woolf")plot(log10(L), log10(v / (n - v)), type ="o", main ="Hill")
Principais linearizações da isoterma de ligação ligante-proteína.
par(mfrow =c(1, 1)) # volta à janela gráfica normal
Desvios da linearidade, por outro lado, são frequentemente utilizados como diagnósticos para modelos que distintos do de homogeneidade de sítios de ligação como acima (heterogeneidade de sítios, criação de sítio, cooperatividade. As equações abaixo descrevem esses modelos, e consideram K, constante de equilíbrio de associação ligante-proteína, como o reverso de Kd, a fim de tornar as expressões mais legíveis:
\[
K = \frac{1}{Kd}
\tag{5}\]
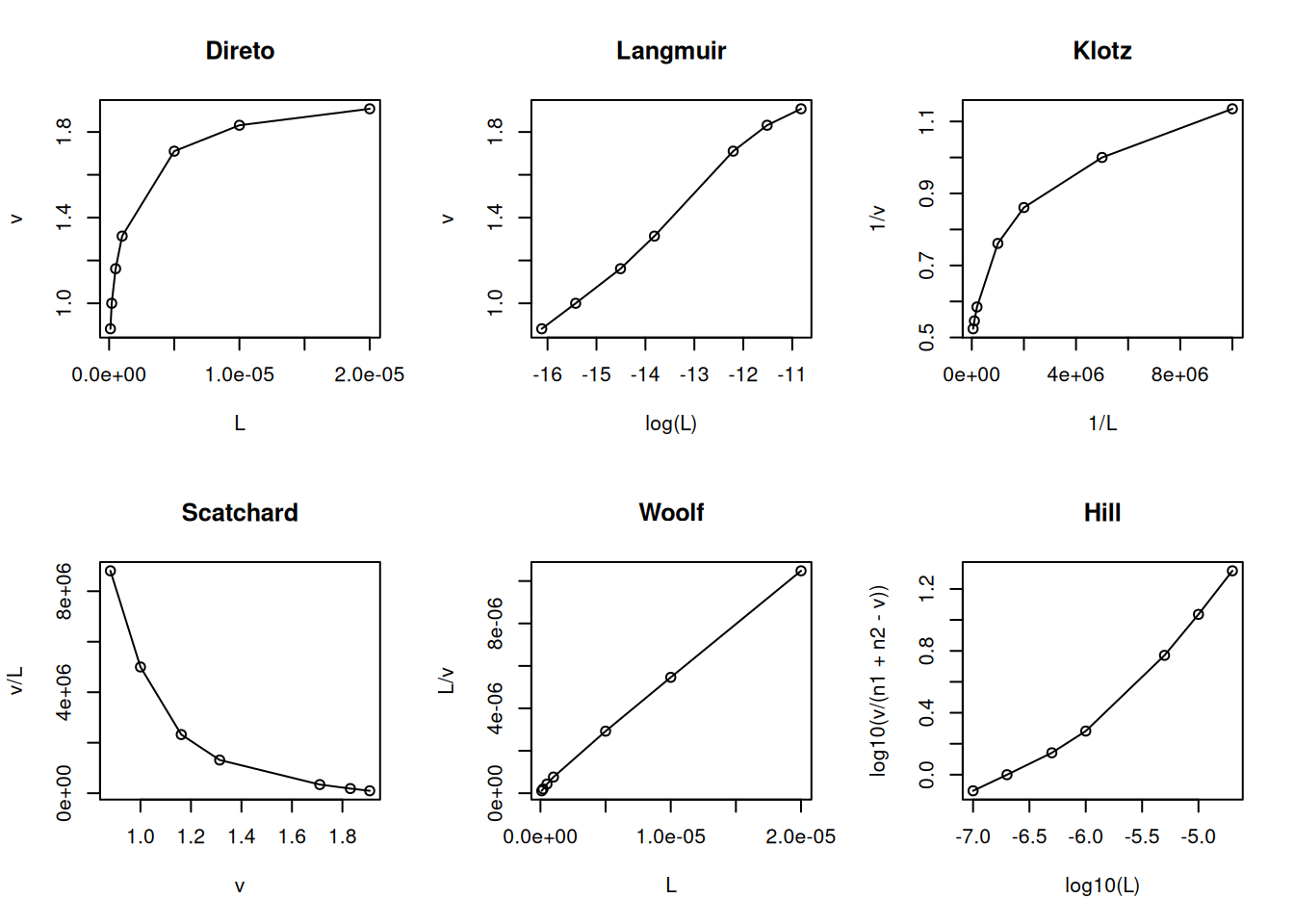
O modelo de heterogeneidade de sítios de ligação pressupõe que haja na proteína mais um sítio com afinidades distintas para o ligante (Dahlquist 1978). Formalmente esse modelo pode ser exemplificado para 2 conjuntos de sítios de ligação, como segue:
O trecho de código abaixo exemplifica o modelo no R, bem como suas principais linearizações diagnósticas.
# Heterogeneidade de sítios de ligaçãoL <-c(0.1, 0.2, 0.5, 1, 5, 10, 20) *1e-6Kd1 <-2e-6n1 <-1Kd2 <-2e-8n2 <-1v <- (n1 * L / (Kd1 + L)) + (n2 * L / (Kd2 + L))par(mfrow =c(2, 3)) # estabelece área de plot pra 6 gráficosplot(L, v, type ="o", main ="Direto")plot(log(L), v, type ="o", main ="Langmuir")plot(1/ L, 1/ v, type ="o", main ="Klotz")plot(v, v / L, type ="o", main ="Scatchard")plot(L, L / v, type ="o", main ="Woolf")plot(log10(L), log10(v / (n1 + n2 - v)), type ="o", main ="Hill")
Modelo e linearizações para heterogeneidade de 2 conjuntos de sítios de ligação
# n1+n2=ntot no Hillpar(mfrow =c(1, 1)) # volta à janela gráfica normal
O modelo de criação de novo sítio - “one-site creator”; (Parsons e Vallner 1978) estabelece uma cooperatividade positiva resultante da produção de novos sítios para o ligante na proteína. Segue o modelo exemplificado e suas linearizações resultantes.
# Criação de novo sítio sob interação com liganteL <-c(0.1, 0.2, 0.5, 1, 5, 10, 20) *1e-6Kd1 <-2e-6n1 <-1Kd2 <-2e-5n2 <-1nH <-0.5v <- (n1 * L *1/ Kd1) / (1+1/ Kd1 * L) + ((n2 *1/ Kd1 *1/ Kd2 * L^2) / (1+1/ Kd1 * L) * (1+1/ Kd2 * L))par(mfrow =c(2, 3)) # estabelece área de plot pra 6 gráficosplot(L, v, type ="o", main ="Direto")plot(log(L), v, type ="o", main ="Langmuir")plot(1/ L, 1/ v, type ="o", main ="Klotz")plot(v, v / L, type ="o", main ="Scatchard")plot(L, L / v, type ="o", main ="Woolf")plot(log10(L), log10(v / (n - v)), type ="o", main ="Hill")
Modelo e linearizações para criação de novo sítio: 1-site creator.
# n1+n2=ntot no Hillpar(mfrow =c(1, 1)) # volta à janela gráfica normal
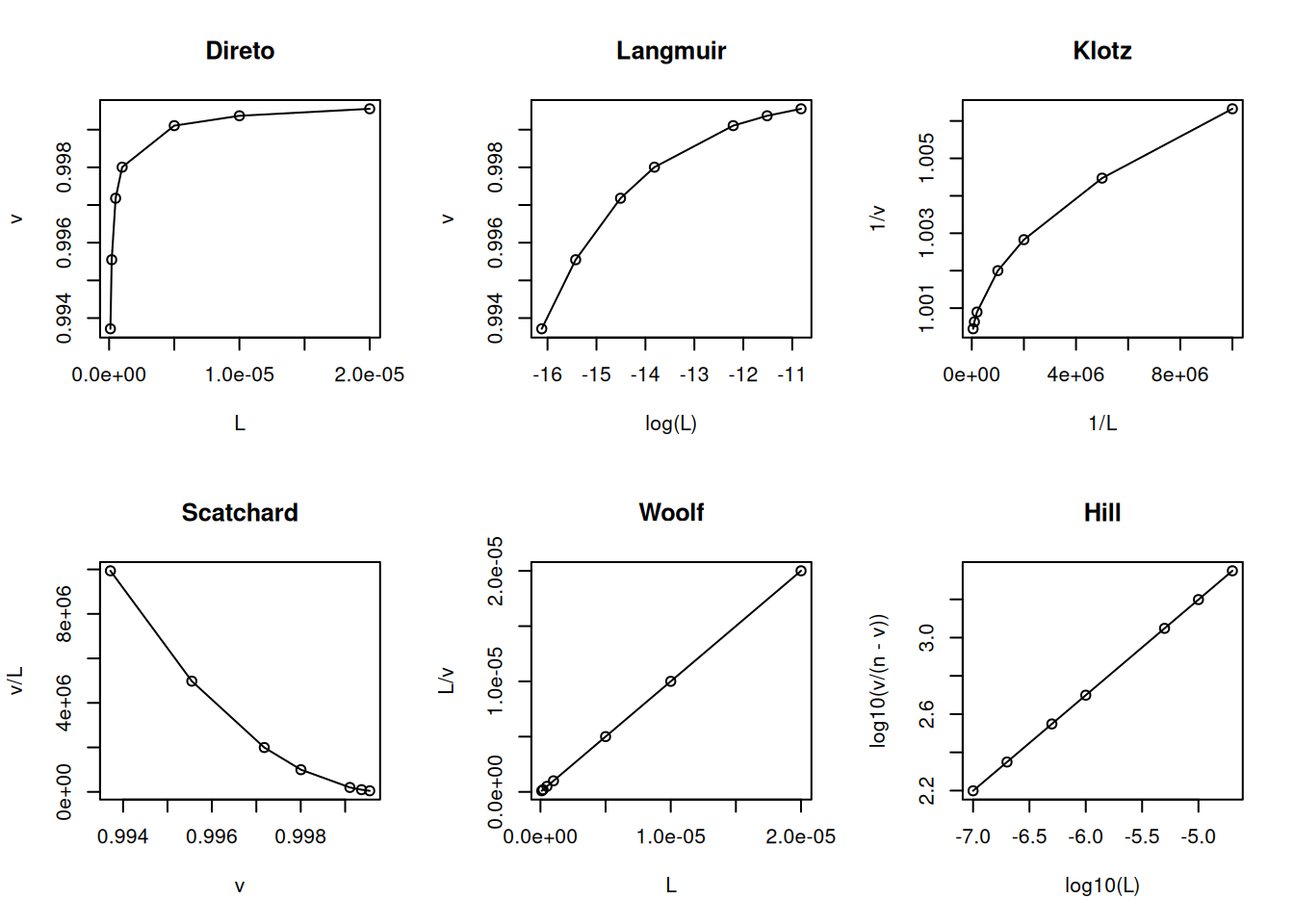
Os modelos de cooperatividade (negativa e positiva) seguem um formalismo similar descrito para a ligação de oxigênio à hemoglobina no capítulos de Proteínas. Na cooperatividade negativa uma segunda molécula de ligante interage com a proteína com menor afinidade:
# Cooperatividade negativa em ligand-bindingL <-c(0.1, 0.2, 0.5, 1, 5, 10, 20) *1e-6Kd <-2e-6n <-1nH <-0.5v <- (n * L^nH / (Kd + L^nH))par(mfrow =c(2, 3)) # estabelece área de plot pra 6 gráficosplot(L, v, type ="o", main ="Direto")plot(log(L), v, type ="o", main ="Langmuir")plot(1/ L, 1/ v, type ="o", main ="Klotz")plot(v, v / L, type ="o", main ="Scatchard")plot(L, L / v, type ="o", main ="Woolf")plot(log10(L), log10(v / (n - v)), type ="o", main ="Hill")
Modelo e linearizações para cooperatividade negativa de sítios de ligação.
# n1+n2=ntot no Hillpar(mfrow =c(1, 1)) # volta à janela gráfica normal
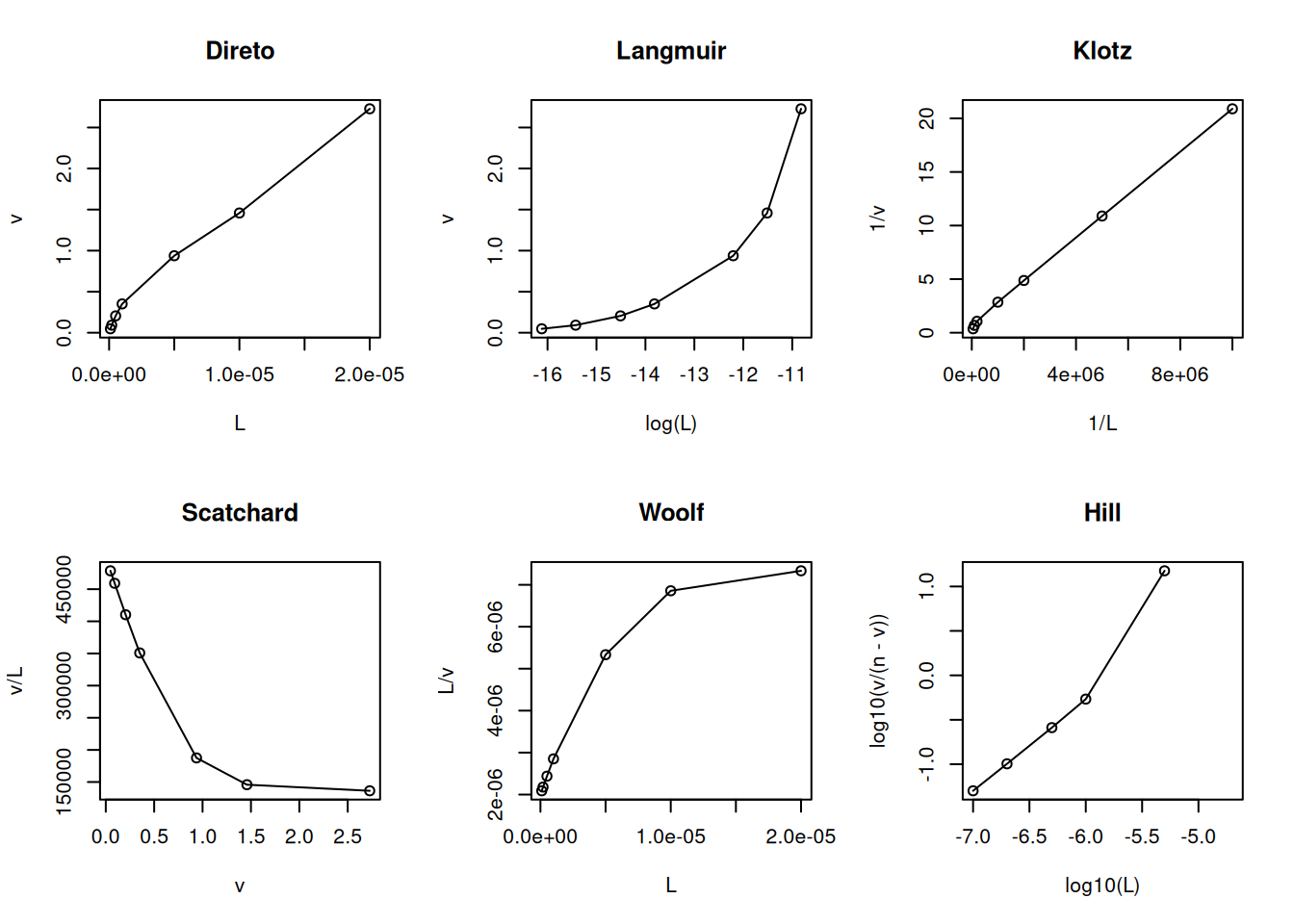
Já na cooperatividade positiva, uma segunda molécula de ligante interage com a proteína com maior afinidade que a primeira molécula (Parsons e Vallner 1978):
# Cooperatividade positiva em ligand bindingL <-c(0.1, 0.2, 0.5, 1, 5, 10, 20) *1e-6Kd <-2e-6n <-1nH <-1.5v <- (n * L^nH / (Kd + L^nH))par(mfrow =c(2, 3)) # estabelece área de plot pra 6 gráficosplot(L, v, type ="o", main ="Direto")plot(log(L), v, type ="o", main ="Langmuir")plot(1/ L, 1/ v, type ="o", main ="Klotz")plot(v, v / L, type ="o", main ="Scatchard")plot(L, L / v, type ="o", main ="Woolf")plot(log10(L), log10(v / (n - v)), type ="o", main ="Hill")
Modelo e linearizações para cooperatividade positiva de sítios de ligação.
# n1+n2=ntot no Hillpar(mfrow =c(1, 1)) # volta à janela gráfica normal
Observe que a inclinação do gráfico de Hill é inferior à unidade para a cooperatividade negativa, e superior a essa, para a cooperatividade positiva, e representa o mesmo parâmetro nH visto no capítulo de Proteinas.
Ainda que sujeito à crítica por sua inconsistência estatística (variável dependente em ambos os eixos), a representação de Scatchard tem sido privilegiada ao longo de décadas como diagnóstico de modelos de interação ligante-proteína. Entre suas vantagens, aloca-se a possibilidade de facilmente distinguir-se o modelo de cooperatividade positiva (aclive) do de heterogeneidade de sítios de ligação (declive abrupto) ou de cooperatividade negativa (declive suave).
Ajuste Não-Linear Em Interação Ligante-Proteína
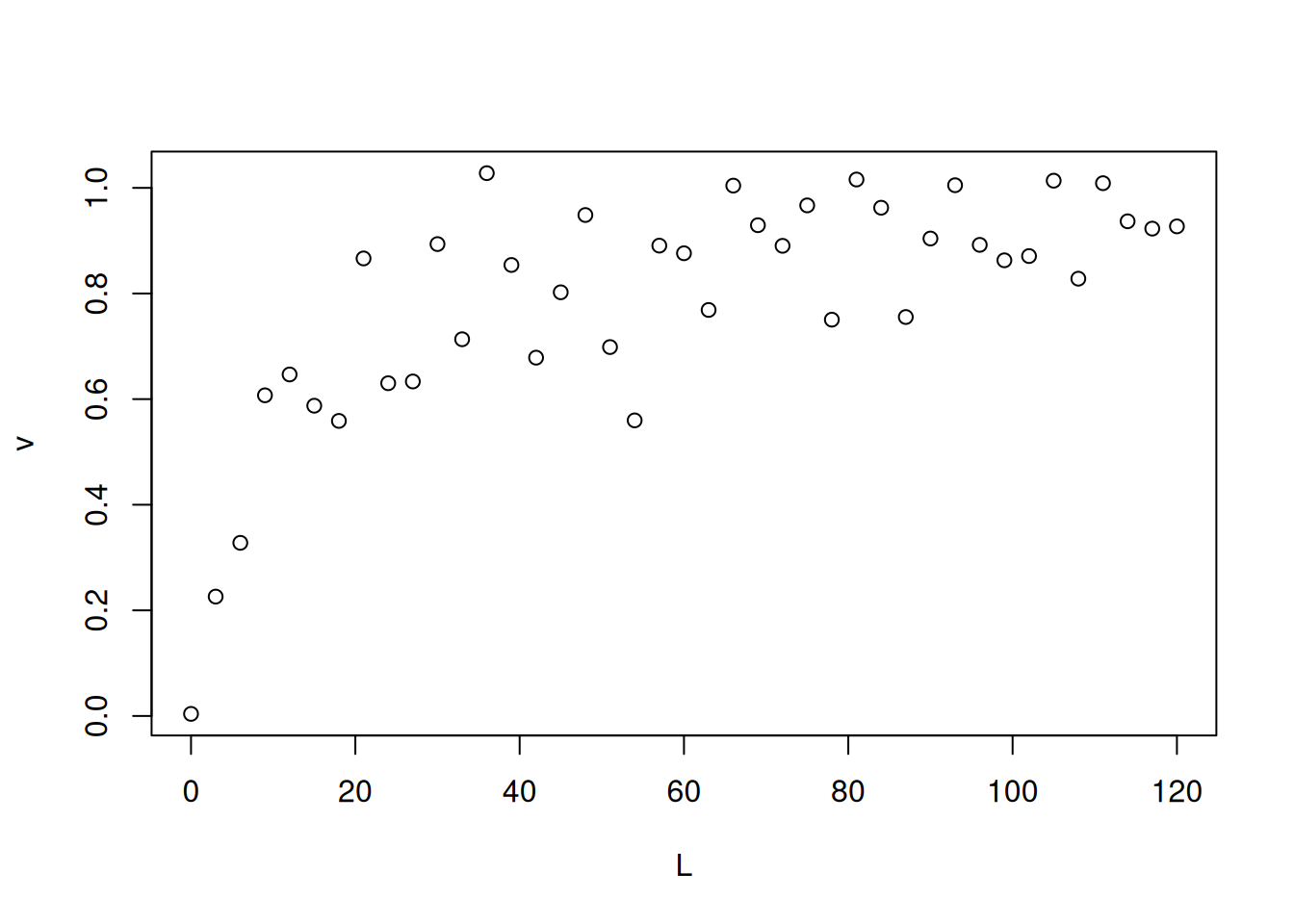
Ajustes diretos da equação não linear dos modelos de interação também podem ser efetuados como fora realizado para a equação de Michaelis-Menten no capítulo de Enzimas. Exemplificando, pode-se simular a obtenção de dados experimentais de binding pelo trecho a seguir, utilizando-se o comando ‘runif’ (random uniform) para geração de sequência aleatória (como fora realizado no capítulo de Enzimas). Para ilustrar, segue a Figura 1.
Observe que o comando ‘rnorm’ adiciona um erro de distribuição normal aos dados. Outra forma para simulação desses é dada abaixo, introduzindo-se o comando ‘runif’ de geração de números aleatórios.
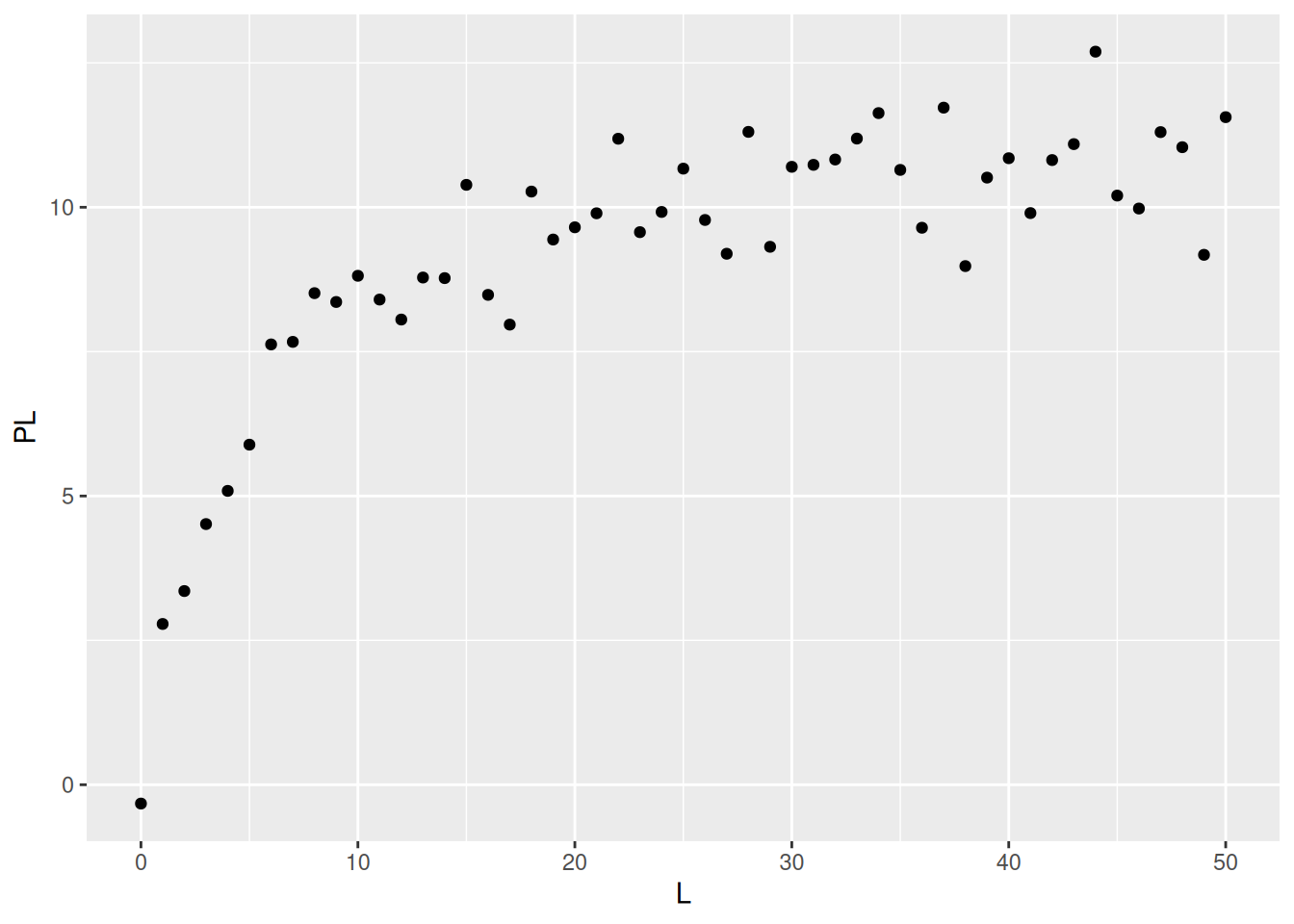
# Simulação de dados de interação bimolecular (1 sítio)# Simulação de dadosset.seed(20160227) # estabelece semente para geração de números aleatóriosL <-seq(0, 50, 1)PL <- ((runif(1, 10, 20) * L) / (runif(1, 0, 10) + L)) +rnorm(51, 0, 1)# 1. runif(n,min,max); quando sem atributos, considera-se min=0 e max=1# 2. rnorm(no. pontos,media,desvio) - erro aleatório de distribuição normalplot(L, PL, xlab ="L", ylab ="PL")
Dados simulados para isoterma de interação bimolecular.
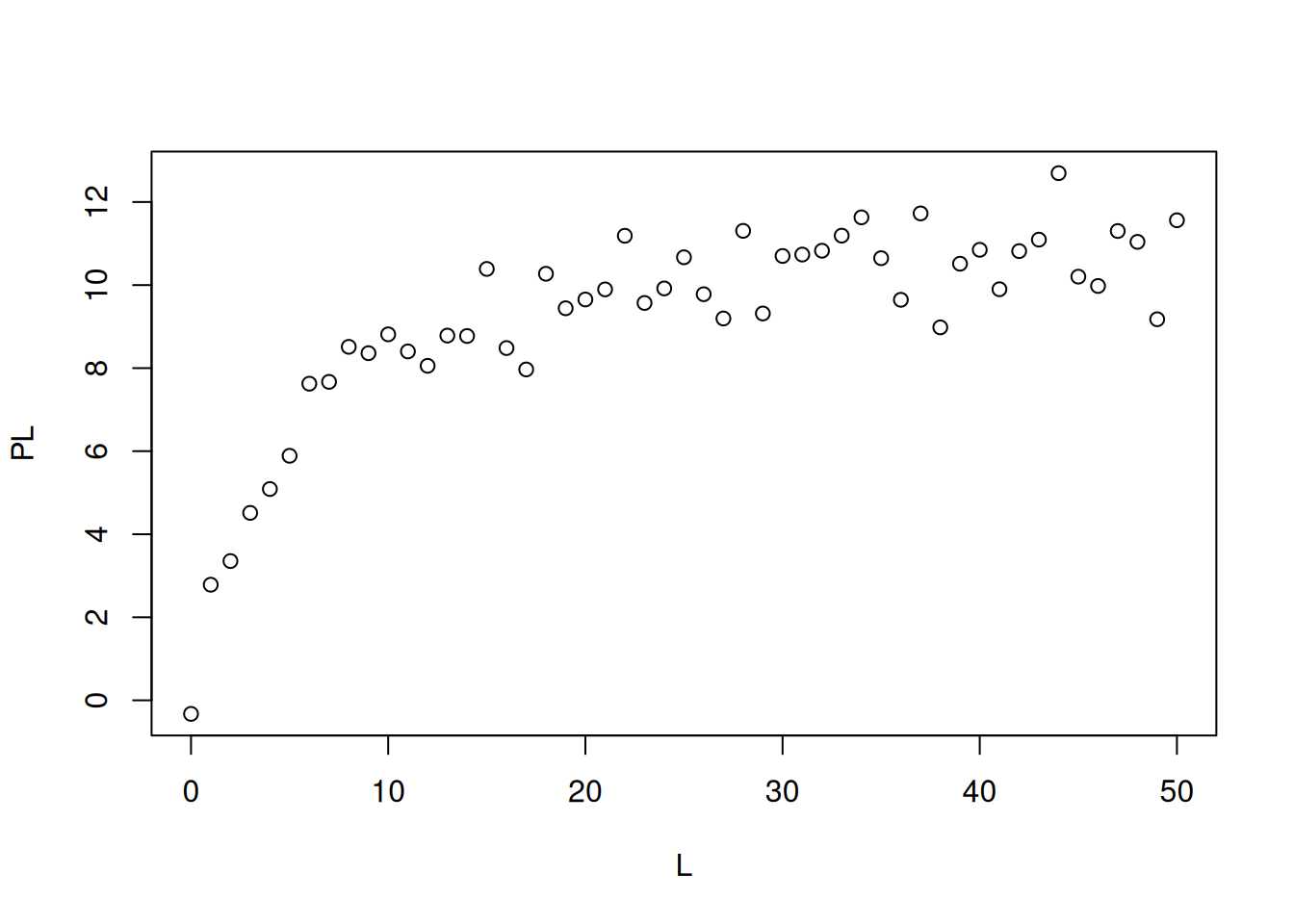
Agora precisamos utilizar o comando ‘nls’ para o ajuste não linear, sobreposição da curva teórica, e tabela estatística de resultados:
# Ajuste não linear em ligand bindingm <-nls(PL ~ n * L / (Kd + L), start =list(n =1, Kd =1))# Coef. de correlaçãocor(PL, predict(m)) # Coeficiente de correlação de Pearson
[1] 0.9496598
# Gráfico de dados e simulaçãoplot(L, PL)lines(L, predict(m), lty =2, col ="red", lwd =3)
summary(m)
Formula: PL ~ n * L/(Kd + L)
Parameters:
Estimate Std. Error t value Pr(>|t|)
n 11.8478 0.2620 45.216 < 2e-16 ***
Kd 4.2778 0.5113 8.366 5.3e-11 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.7826 on 49 degrees of freedom
Number of iterations to convergence: 6
Achieved convergence tolerance: 1.554e-06
Sistemas Gráficos no R
A última curva de simulação obtida o foi junto à biblioteca padrão para manipulação de gráficos da instalação do ‘R’, Graphics. Trata-se um conjunto de funções amplo também utilizado por vários outros pacotes do ambiente. Contudo, existem no ‘R’ diversas outras bibliotecas para elaboração de gráficos, dentre os quais vale destacar o Lattice, também incluido na instalação padrão, e o ggplot2. Ambos os sistemas geram resultados com melhor estética e flexibilidade gráfica que a biblioteca Graphics padrão, e possuem empregos e semânticas distintas entre si.
O sistema Lattice(Sarkar 2008) é baseado no sistema Trellis para representação gráfica de dados multivariados. Sua força está na representação de dados em paineis contendo subgrupos e, embora tenha sintaxe menos intuitiva e por vezes mais elaborada que o pacote Graphics, produz um grafismo superior a esse com poucos cliques de teclado. De modo geral, o Lattice produz o gráfico dentro do próprio algoritmo, de modo diferente aos sistemas Graphics (pode-se acumular linhas sucessivas de modificação do gráfico) ou ggplot2.
Por outro lado a biblioteca ggplot2 é baseada na gramática de gráficos (Wickham 2011), e produz o gráfico utilizando uma única linha de comando que combina camadas sobrepostas, de modo similar à aplicativos de manipulação de imagens (ex: Inkscape, Gimp, Corel Draw, Photoshop). Dessa forma é possível alterar cada ítem do gráfico em suas camadas específicas (tema, coordenadas, facets, estatísticas, geometria, estética, dados). Exemplificando o resultado gráfico da curva de simulação acima de binding para Lattice e ggplot2:
# Os sistemas lattice e ggplot2# Simulação de dadosset.seed(20160227) # estabelece semente para geração de números aleatóriosL <-seq(0, 50, 1)PL <- ((runif(1, 10, 20) * L) / (runif(1, 0, 10) + L)) +rnorm(51, 0, 1)# 1. runif(n,min,max); quando sem atributos, considera-se min=0 e max=1# 2. rnorm(no. pontos,media,desvio) - erro aleatório de distribuição normal# Produção do gráfico com sistema Latticelibrary(lattice)xyplot(PL ~ L)
# Produção do gráfico com sistema ggplot2library(ggplot2)qplot(L, PL)
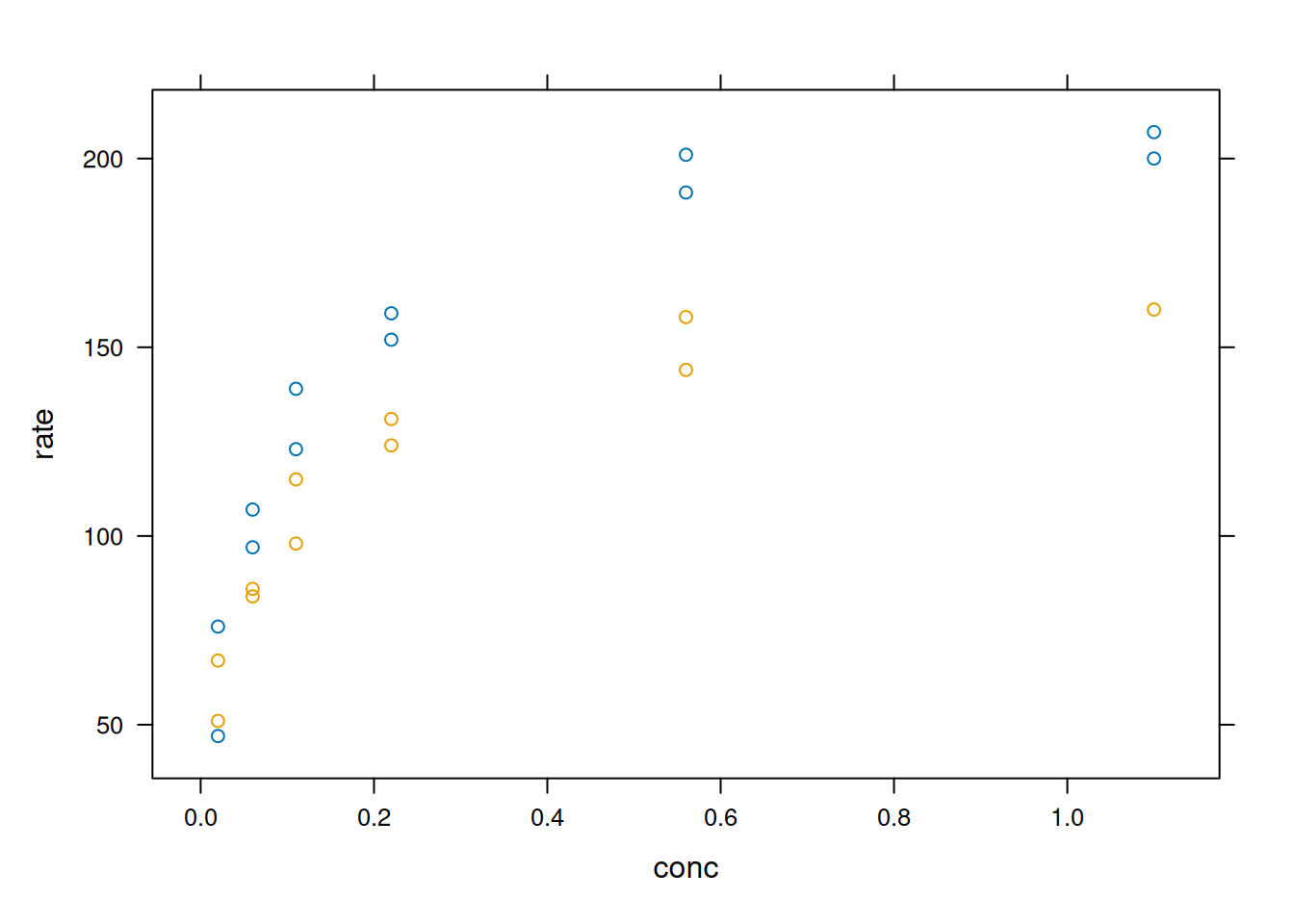
Como a percepção de peculiaridades dos sistemas gráficos se revela melhor com dados mais elaborados, as ilustrações a seguir utilizarão o conjunto de dados ‘Puromycin’, que integra a biblioteca ‘datasets’ do ‘R’. Os dados apresentam a velocidade de reação enzimática sobre um substrato em células tratadas e não tratadas com puromicina.
O código abaixo retorna a plotagem, ajuste não linear e resultados obtidos com a biblioteca Graphics padrão, e foi extraído do própria documentação do dataset.
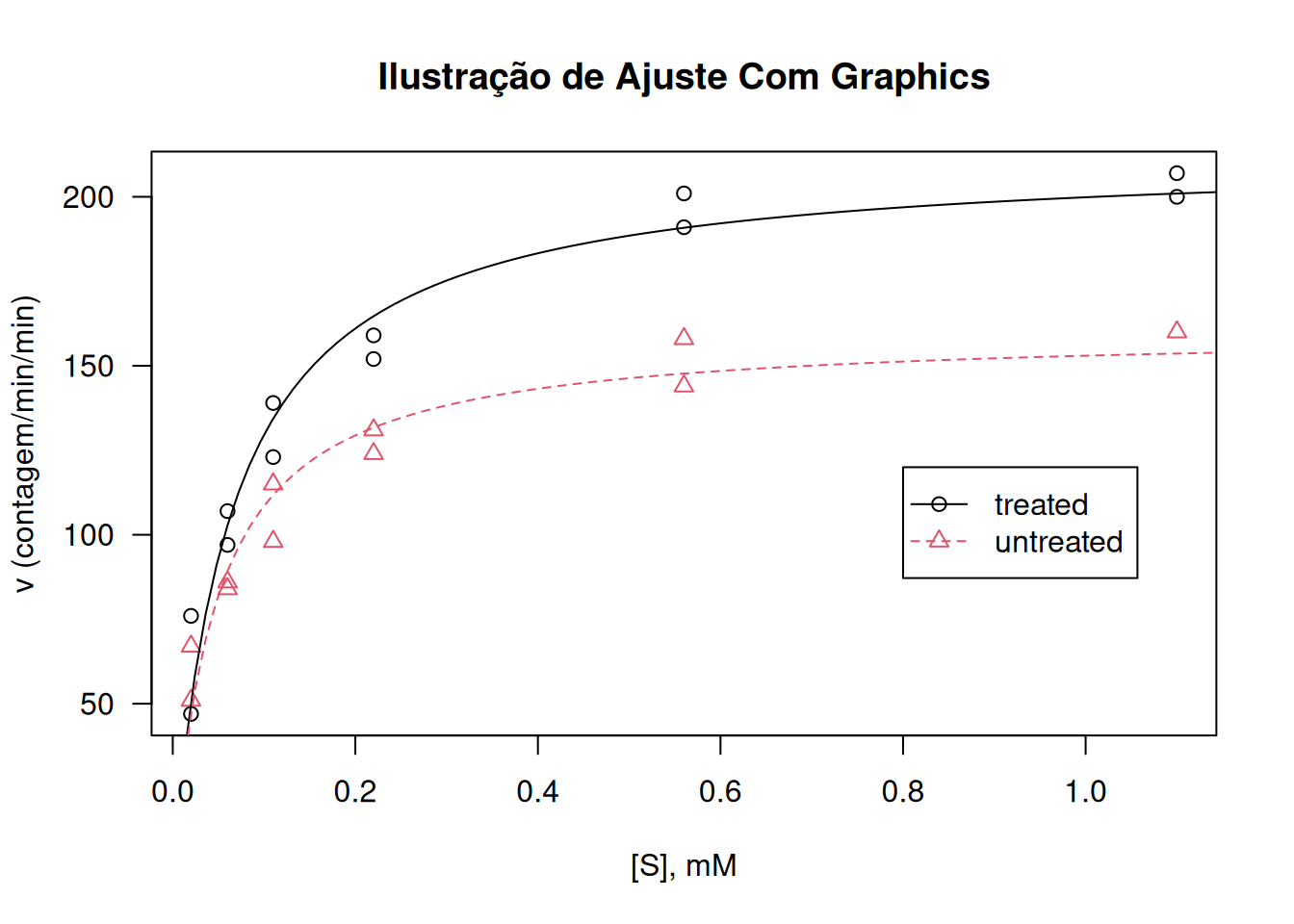
library(datasets)# O sistema base `graphics`plot(rate ~ conc,data = Puromycin, las =1,xlab ="[S], mM",ylab ="v (contagem/min/min)",pch =as.integer(Puromycin$state),col =as.integer(Puromycin$state),main ="Ilustração de Ajuste Com Graphics")## Ajuste da equação de Michaelis-Mentemfm1 <-nls(rate ~ Vm * conc / (K + conc),data = Puromycin,subset = state =="treated",start =c(Vm =200, K =0.05))fm2 <-nls(rate ~ Vm * conc / (K + conc),data = Puromycin,subset = state =="untreated",start =c(Vm =160, K =0.05))summary(fm1)
Formula: rate ~ Vm * conc/(K + conc)
Parameters:
Estimate Std. Error t value Pr(>|t|)
Vm 2.127e+02 6.947e+00 30.615 3.24e-11 ***
K 6.412e-02 8.281e-03 7.743 1.57e-05 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 10.93 on 10 degrees of freedom
Number of iterations to convergence: 5
Achieved convergence tolerance: 8.768e-06
summary(fm2)
Formula: rate ~ Vm * conc/(K + conc)
Parameters:
Estimate Std. Error t value Pr(>|t|)
Vm 1.603e+02 6.480e+00 24.734 1.38e-09 ***
K 4.771e-02 7.782e-03 6.131 0.000173 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 9.773 on 9 degrees of freedom
Number of iterations to convergence: 5
Achieved convergence tolerance: 4.473e-06
## Adição de linhas de ajuste ao plotconc <-seq(0, 1.2, length.out =101)lines(conc, predict(fm1, list(conc = conc)), lty =1, col =1)lines(conc, predict(fm2, list(conc = conc)), lty =2, col =2)legend(0.8, 120, levels(Puromycin$state),col =1:2, lty =1:2, pch =1:2)
Plotagem e análise com graphics.
O sistema ggplot2, por sua vez, exige que os comandos sejam elencados em camadas justapostas intercaladas com o sinal “+”, como segue:
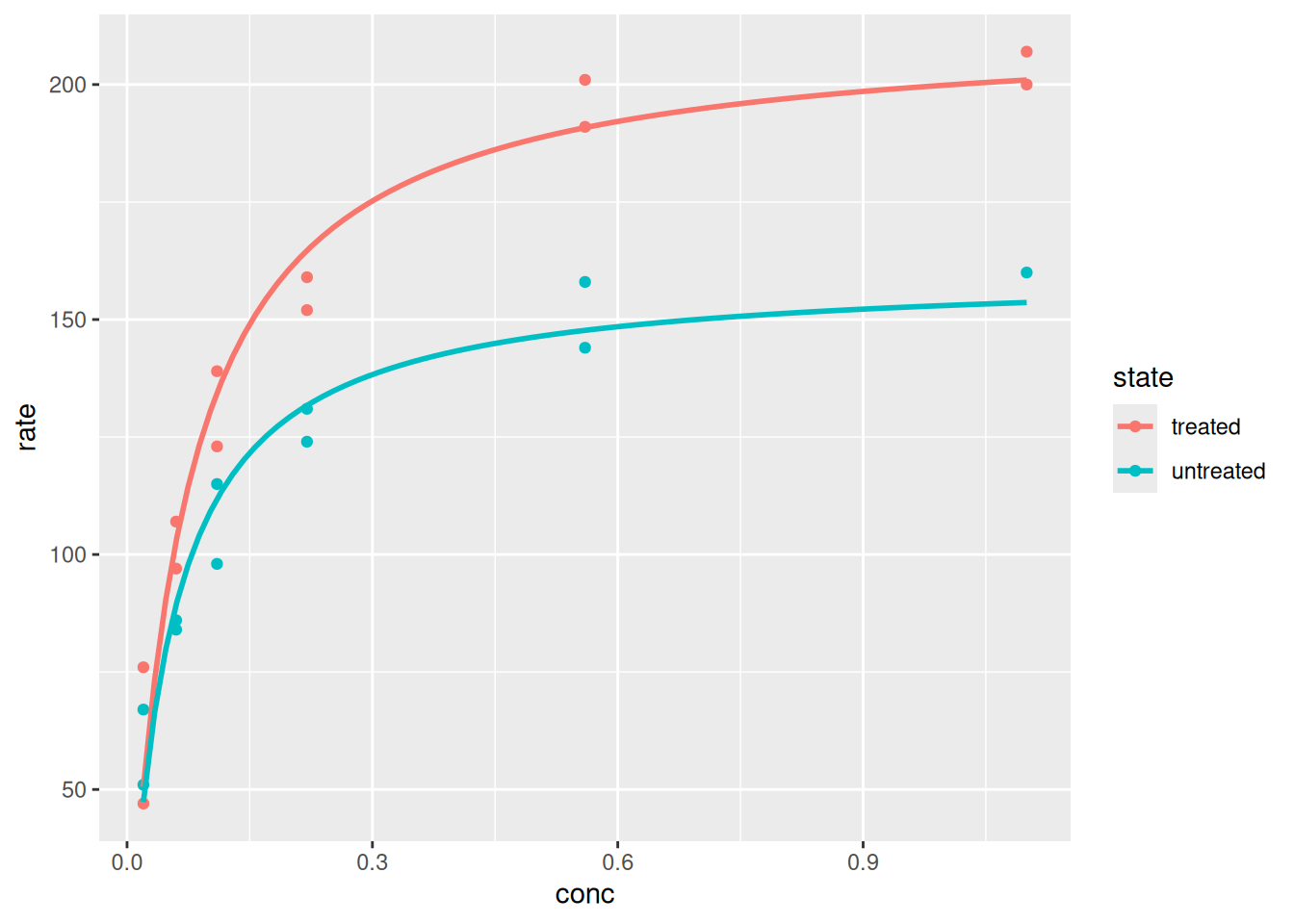
# Gráfico e análise com ggplot2library(datasets)p <-ggplot(data = Puromycin, aes(conc, rate, color = state)) +geom_point() +geom_smooth(method ="nls",formula = y ~ Vm * x / (Km + x),method.args =list(start =list(Vm =200, Km =0.1)),se =FALSE ) # expressão que define o plotp # variável que apresenta o plot
Plotagem e análise com ggplot2.
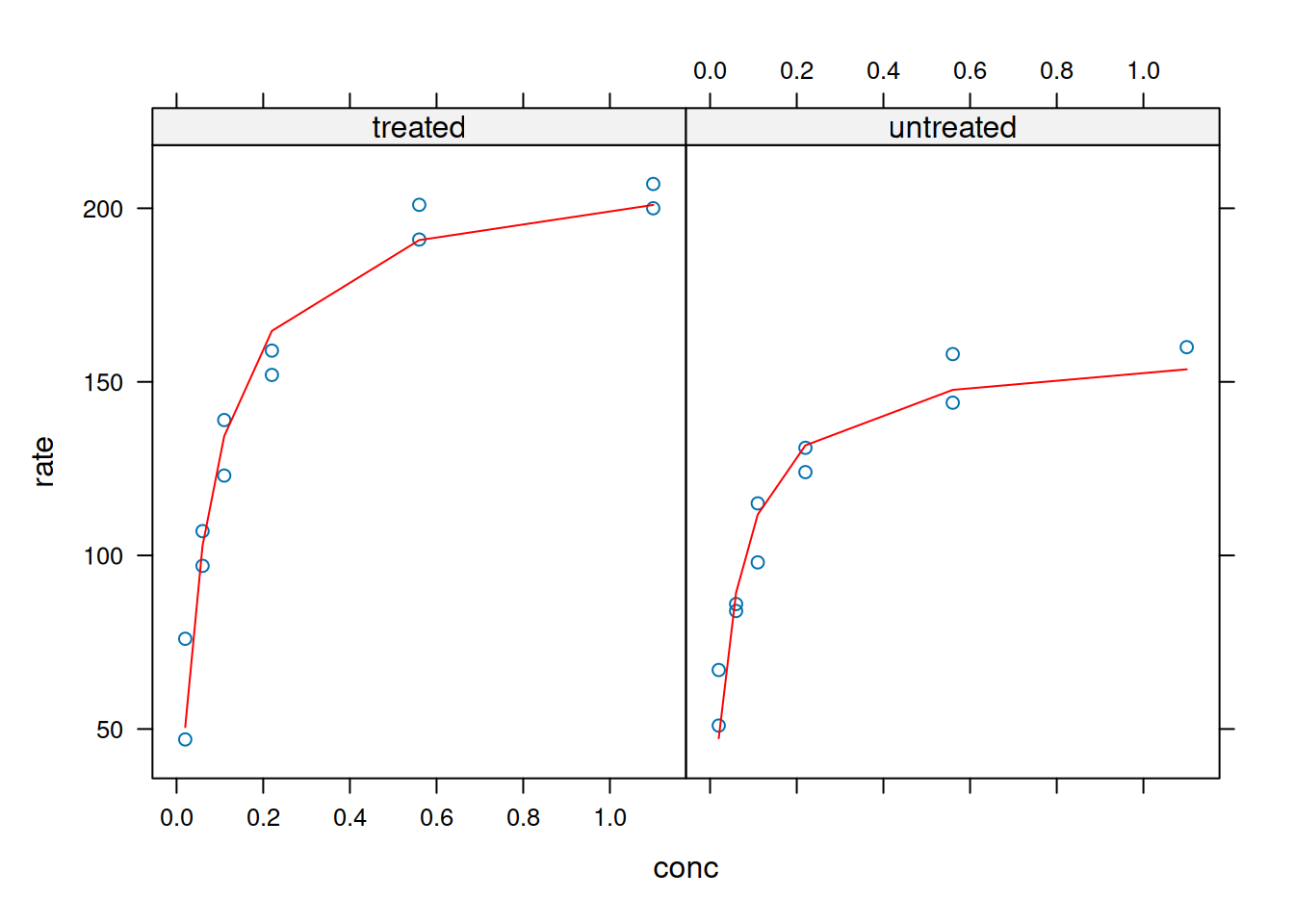
Perceba o menor número de instruções do script para a produção do gráfico. Além disso, e diferente do Graphics, ggplot2 permite adicionar camadas à linha de comandos principais, e apresentar os dados multivariados em paineis (funções ‘facet_grid’ e ‘facet_wrap’), sem a necessidade de se utilizar o comando ‘mfrow’ ou ‘mfcol’ visto no capítulo, como segue:
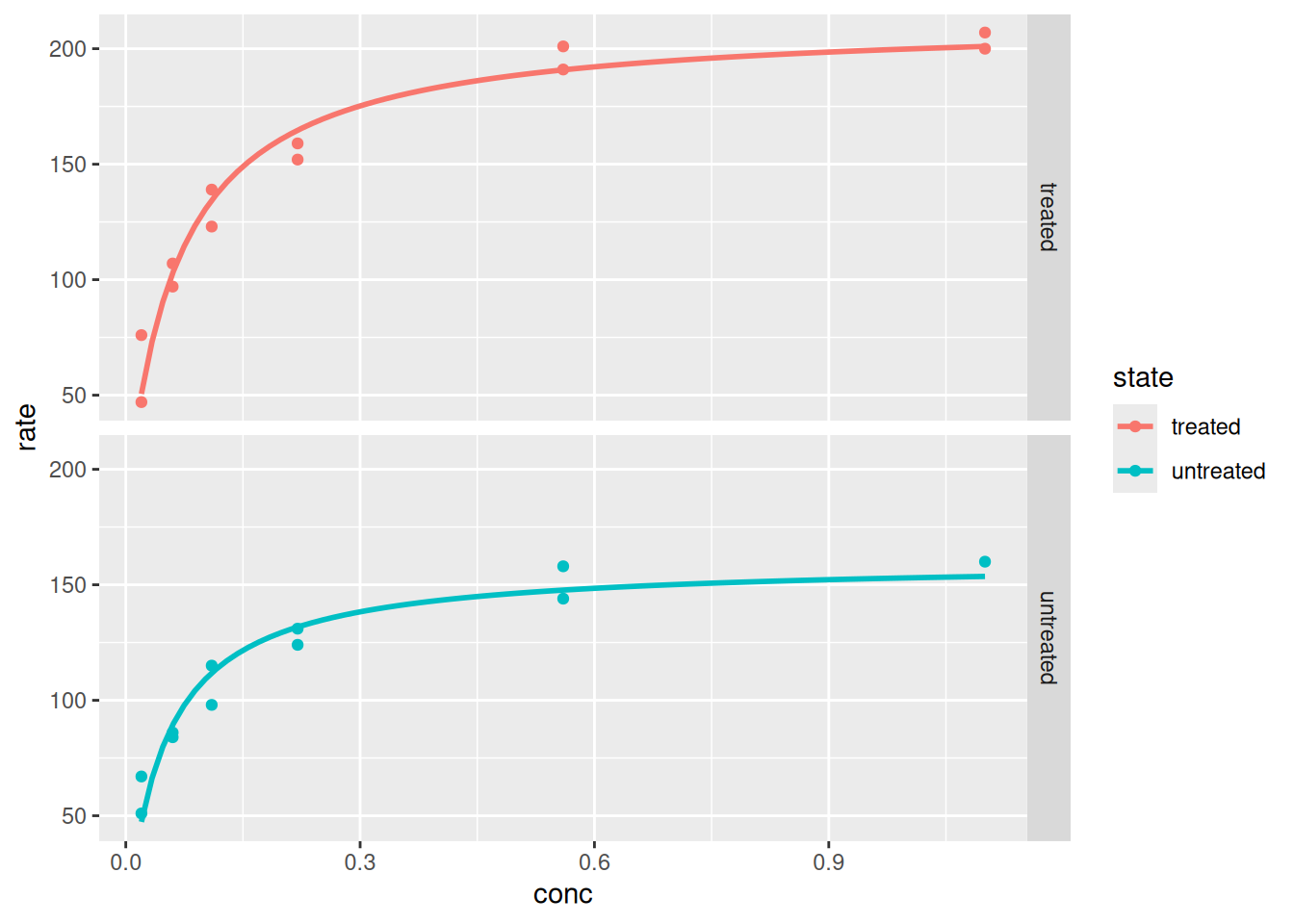
p +facet_grid(rows =vars(state))
Plotagem e análise com ggplot2 - paineis (faceting).
Lattice também possui uma economia de instruções em relação à Graphics. Não considerando o ajuste estatístico, os grupos podem ser apresentados simplesmente utilizando-se a fórmula:
\[
xyplot(y \sim x~|~ groups = z)
\tag{7}\]
library(lattice)xyplot(rate ~ conc, data = Puromycin, groups = state)
E para a representação dos ajustes não lineares:
# Um gráfico com dataset para latticelibrary(nlme)n1 <-nlsList(rate ~ Vmax * conc / (Km + conc) | state, data = Puromycin, start =list(Vmax =200, Km =0.1))summary(n1)xyplot(rate ~ conc,groups = state, data = Puromycin) +layer(panel.curve(Vmax[1] * x / (Km[1] + x), col =1),data =as.list(coef(n1)) ) +layer(panel.curve(Vmax[2] * x / (Km[2] + x), col =2),data =as.list(coef(n1)) )
A biblioteca Lattice também permite a apresentação em paineis; diferente de ggplot2, contudo, o gráfico é gerado algoritmicamente, sem a sobreposição de comandos:
# Gráfico e análise não linear com lattice library(nlme) # pacote quer permite regressão não linear com subgruposnonlinLatt <-nlsList(rate ~ Vmax * conc / (Km + conc) | state, start =list(Vmax =200, Km =0.1), data = Puromycin)summary(nonlinLatt)
Call:
Model: rate ~ Vmax * conc/(Km + conc) | state
Data: Puromycin
Coefficients:
Vmax
Estimate Std. Error t value Pr(>|t|)
treated 212.6836 6.608088 32.18535 3.241151e-11
untreated 160.2800 6.896011 23.24242 1.384612e-09
Km
Estimate Std. Error t value Pr(>|t|)
treated 0.06412110 0.007876774 8.140529 0.0000156514
untreated 0.04770812 0.008281147 5.761052 0.0001727056
Residual standard error: 10.40003 on 19 degrees of freedom
Plotagem e análise com Lattice - ajuste externo e paineis.
Das muitas diferenças que os pacotes ggplot2 e Lattice apresentam, há uma que vale a pena ressaltar. Como visto acima, o ggplot2 realiza o ajuste não linear dentro da linha de comandos de geração do gráfico, ao passo que o Lattice permite fazê-lo fora da linha. Isso é inerente do ggplot2, uma biblioteca desenhada para a produção de gráficos, e não para análises computacionais. Dessa forma o algoritmo que permite o ajuste não linear pelo ggplot2, ainda que seja o mesmo ‘nls’ já trabalhado, não expressa seus resultados explicitamente (embora haja formas de “pescá-los” utilizando-se outros pacotes).
De certa forma, ainda que o Lattice exija uma curva de aprendizado menos intutiva, ele permite que se utilize os resultados estatísticos obtidos anteriormente para inclusão no algoritmo de plotagem. Isso é vantajoso quando se deseja outros algoritmos estatísticos para ajuste, como acima, ou mesmo sua flexibilização, além do ‘nls’ incluido em ggplot2. Não obstante, o Lattice também permite que se inclua a linha de ajuste dentro do próprio algoritmo, como abaixo:
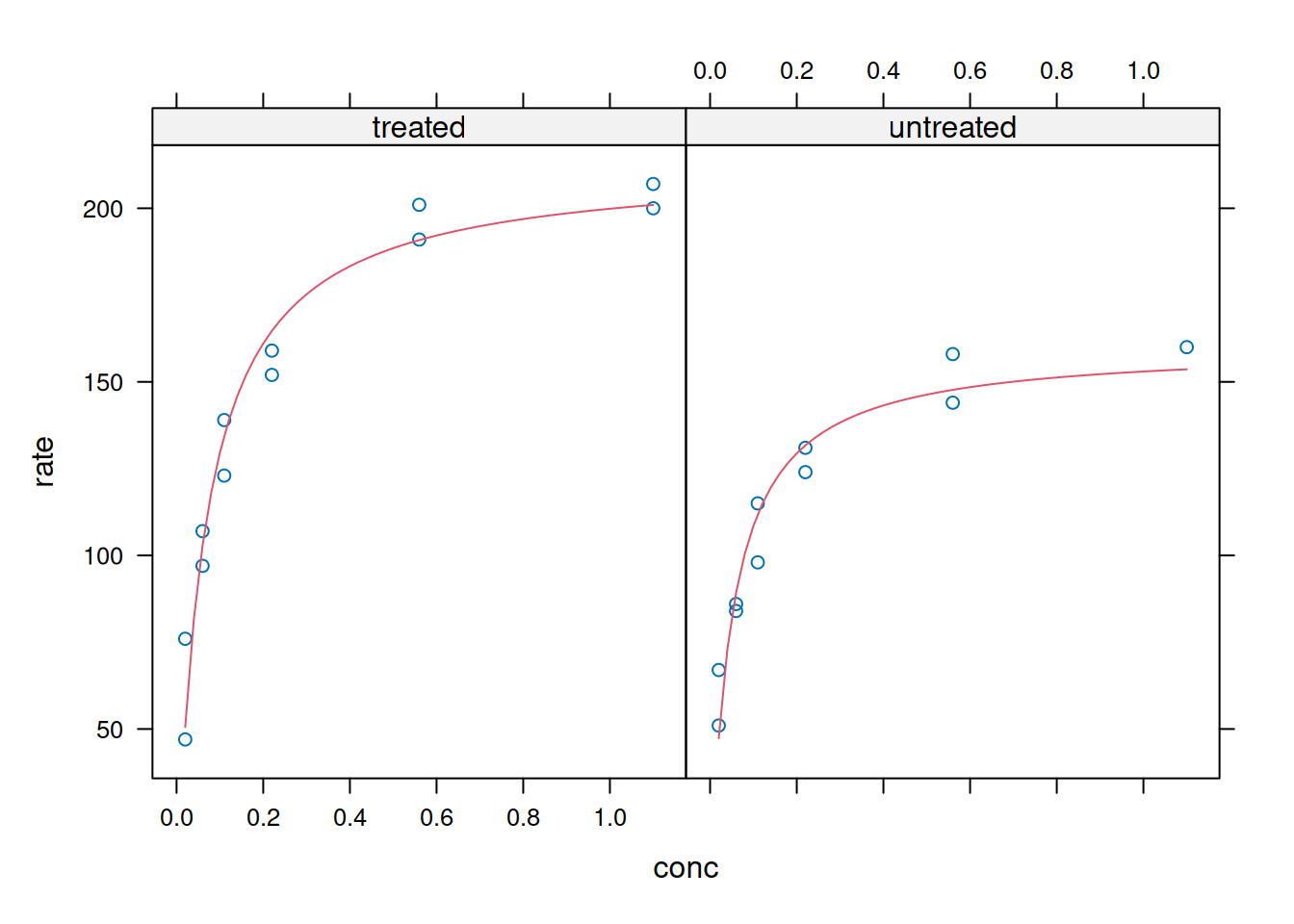
# Ajuste não linear em paineis (Lattice)xyplot(rate ~ conc | state,data = Puromycin,panel =function(x, y, ...) {panel.xyplot(x, y, ...) n3 <-nls(y ~ Vmax * x / (Km + x), data = Puromycin, start =list(Vmax =200, Km =0.1))panel.lines(seq(0.02, 1.1, 0.02), predict(n3, newdata =data.frame(x =seq(0.02, 1.1, 0.02))),col.line =2) },xlab ="conc", ylab ="rate")
Plotagem e análise com Lattice - ajuste interno e paineis.
Solução Numérica Para o Equilíbrio de Complexos Ligante-Proteína
Como visto no capítulo de Aminoácidos, por vezes uma solução numérica pode ser empregada quando a solução analítica não converge, ou quando estamos diante de uma equação mais complexa. Nesse sentido a formação de complexos de que trata este subtítulo pode também ser tratada por uma solução numérica.
Usualmente o tratamento dado para a solução numérica envolve encontrar as raízes de uma equação ou sistema de equações, ou seja:
Assim, tem-se um sistema de equações lineares nos parâmetros ([P] livre, [L] livre, e complexo [PL]) que pode ser solucionado pelo R por diversas maneiras, uma das quais pela função de minimização rootSolve:
# Cálculo de L, P, e PL em interação biomolecular para 1 conjunto de sítios# de mesma afinidadelibrary(rootSolve)Pt <-1Lt <-10Kd <-4# Modelomodel <-function(x) c(x[1] + (x[1] * x[2]) / Kd - Pt, x[2] + (x[1] * x[2]) / Kd - Lt, Pt - x[1] - x[3])# o modelo acima deve conter uma lista de equações cuja igualdade é zero, # ou seja, f(x)=0(ss <-multiroot(model, c(1, 1, 1))) # comando de execução do
Dessa forma os valores resultantes (f.root) quando [Lt] = 10 são apresentados como [P] = 0,3, [L] = 9,3 e [PL] = 0,7.
Outras soluções numéricas permitem um maior controle sobre o algoritmo empregado, tais como a função optim do R (limites de busca da solução, emprego de vetores, por ex). Para isso será exemplificado a mesma situação acima, embora apresentando uma variação do formalismo que relaciona P, L e PL :
Aplicando-se o algoritmo de minimização optim do R:
# Cálculo de L, P, PL em interação para 1 sítiomodel2 <-function(x, Pt, Lt, K) { L <- x[1] P <- x[2] PL <- x[3] (Pt - P - PL)^2+ (Lt - L - PL)^2+ (P * L - Kd * PL)^2} # declaração da funçãoPt <-1Lt <-10Kd <-4# parâmetros da funçãosol2num <-optim(c(0.5, 1, .5), model2, method ="L-BFGS-B", lower =c(0, 0, 0), upper =c(Lt, Pt, Pt), Pt = Pt, Lt = Lt)# método BFGS permite bounds (lower, upper)sol2num$par # LF, PF, PL calculados
[1] 9.3007349 0.3007355 0.6992652
Perceba que são os mesmos resultados anteriores, embora com maior controle da solução. Agora pode-se utilizar essa minimização para criar um vetor de soluções para as três quantidades, como segue:
# Declaração da funçãobind1 <-function(x, Pt, Lt, Kd) { L <- x[1] P <- x[2] PL <- x[3] (Pt - P - PL)^2+ (Lt - L - PL)^2+ (P * L - Kd * PL)^2}# Parâmetros da funçãoPt <-1Lt <-c(5, 10, 20)Kd <-4# Minimização (parâmetros para que a função acima dê zero)y <-function(i) {optim(c(1, 1, 1), bind1,method ="L-BFGS-B", lower =c(0, 0, 0), upper =c( Lt[i], Pt, Pt ), Lt = Lt[i], Pt = Pt, Kd = Kd )}# Resultados em matrizypar <-function(i) y(i)$paryp <-matrix(nrow =length(Lt), ncol =2+length(Kd),byrow = T)for (i in1:length(Lt)) yp[i, ] <-y(i)$parcolnames(yp) <-c("L", "P", "PL")rownames(yp) <-c("5", "10", "20")yp
L P PL
5 4.472136 0.4721359 0.5278640
10 9.300736 0.3007344 0.6992634
20 19.172624 0.1726180 0.8273844
Cinética de Interação Ligante-Proteína e Solução Numérica
Sob o mesmo princípio da solução numérica apresentada no ítem anterior para o equilíbrio da interação ligante-proteína, o R permite solução de mesma natureza para a cinética da formação dos complexos, ou seja, os teores de P, L e PL observados no tempo. Nesse caso pode-se desenvolver outras relações a partir da Equação 1. Tomando-se por base que no equilíbrio as taxas cinéticas de k\(_{on}\) e k\(_{off}\) se igualam (steady-state), pode-se relacionar algumas equações diferenciais para a associação, bem como para a dissociação dos complexos:
A solução para esse sistema final de equações diferenciais (taxas líquidas) envolve resolver a variação de cada quantidade (\(\Delta\)x) num determinado intervalo de tempo (\(\Delta\)t), tal que:
\[
\Delta x=f(x)*\Delta t
\tag{16}\]
Para isso é necessário utilizar uma biblioteca do R que permita a solução de um sistema de equações diferenciais. Entre as muitas soluções (odeintr, pracma, rODE), o emprego da biblioteca deSolve, que utiliza uma função para integração do sistema por algoritmo de Runge-Kutta de 4a. ordem:
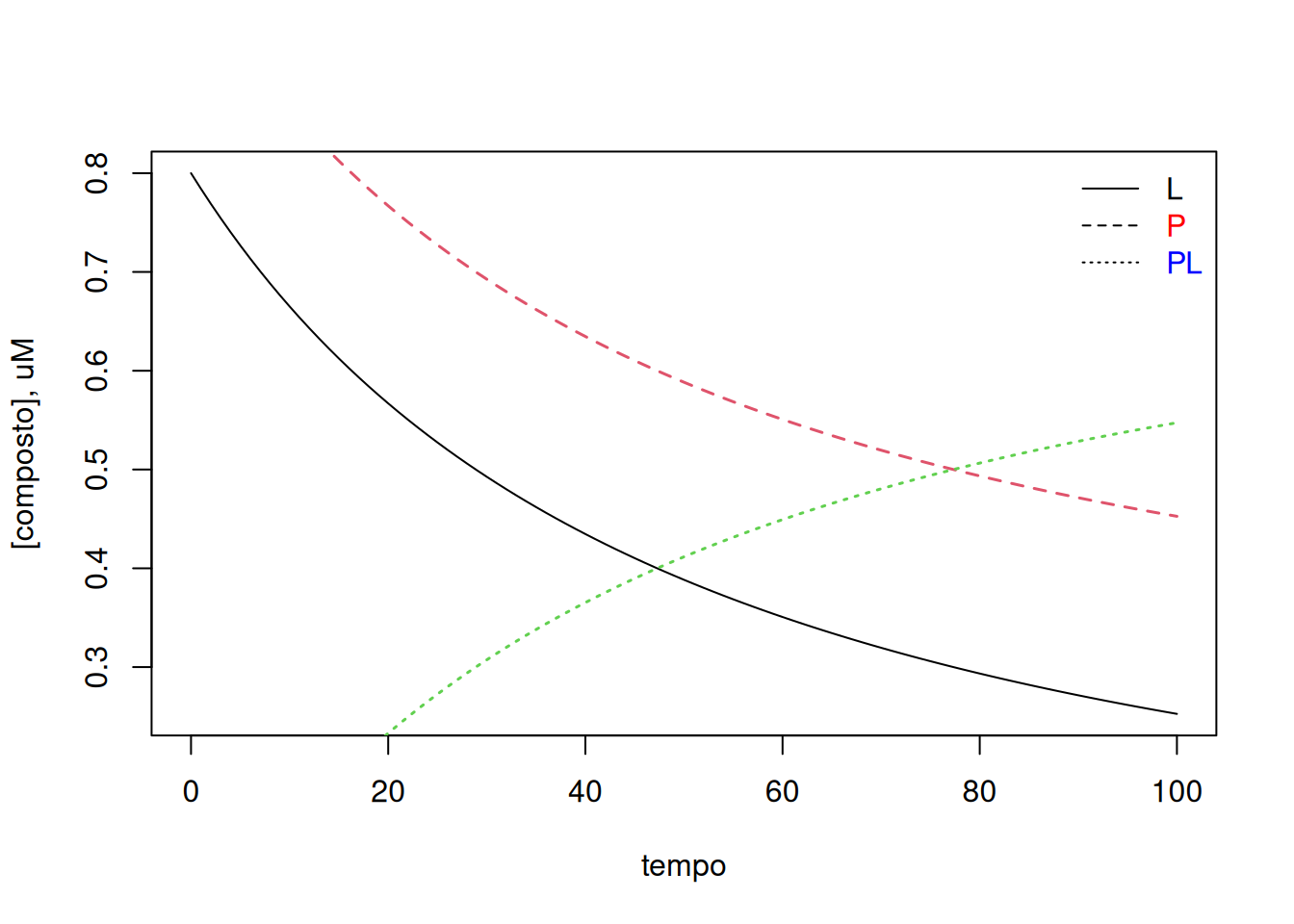
# Cinética de interação ligante-protéina para 1 conjunto de sítioslibrary(deSolve)# Condições experimentaistempo <-seq(0, 100) # intervalo de tempoparms <-c(kon =0.02, koff =0.001) # parâmetros do estado estacionário # da interação (uM^-1*s^-1 e s^-1, respectivamente)val.inic <-c(L =0.8, P =1, PL =0) # valores iniciais, uM# Integração do sistema por Runge-Kutta de 4a. ordemsolNumKin <-function(t, x, parms) {# definição da lista de parâmetros L <- x[1] # ligante P <- x[2] # proteína PL <- x[3] # complexowith(as.list(parms), {# definição da lista de equações diferenciais dL <--kon * L * P + koff * PL dP <--kon * L * P + koff * PL dPL <- kon * L * P - koff * PL res <-c(dL, dP, dPL)list(res) })}sol.rk4 <-as.data.frame(rk4( val.inic, tempo, solNumKin, parms)) # rotina para Runge-Kutta 4a. ordem# Gráficoplot(sol.rk4$time, sol.rk4$L, type ="l", xlab ="tempo", ylab ="[composto], uM")legend("topright", c("L", "P", "PL"), text.col =c("black", "red", "blue"), bty ="n", lty =c(1, 2, 3))lines(sol.rk4$time, sol.rk4$P, type ="l", lty =2, col =2, lwd =1.5)lines(sol.rk4$time, sol.rk4$PL, type ="l", lty =3, col =3, lwd =1.5)
Teores de ligante e proteína livres (L e P), bem como do complexo PL apresentados ao longo do tempo de acordo com o método Runge-Kutta de 4a. ordem para solução de equações diferenciais.