3 Seleção de Composto Líder para Alvos Enzimáticos - LH-Toolkit
Diversas são as etapas que envolvem o desenvolvimento de um fármaco ou composto bioativo. Entre essas, é habitual começar com uma triagem inicial (screening) envolvendo ensaios de alto rendimento (high throughput screening, HTS), além de experimentos para curvas de dose-resposta, esses visando a determinação de IC\(_{50}\) e seleção de inibidores com melhor potencial. O padrão de inibição é caracterizado pelos modelos clássicos observados na Seção 2.1, partindo-se para validação, seletividade, e seleção de um composto líder baseada em critérios (ex: potência adequada, seletividade para a enzima-alvo, eficiência do ligante, solubilidade e estabilidade química, e otimização estrutural). A esses estudos iniciais seguem estudos biofísicos complementares (ex: SPR-Surface Plasmon Resonance, ITC-Isothermal Titration Calorimetry, cristalografia de raios-X, NMR-Nuclear Magnetic Resonance) para quantificação de parâmetros estruturais e termodinâmicos, além de ensaios celulares para avaliar a permeabilidade celular, toxicidade e efeito farmacológico esperado (drug-likeness).
3.1 LH-Toolkit
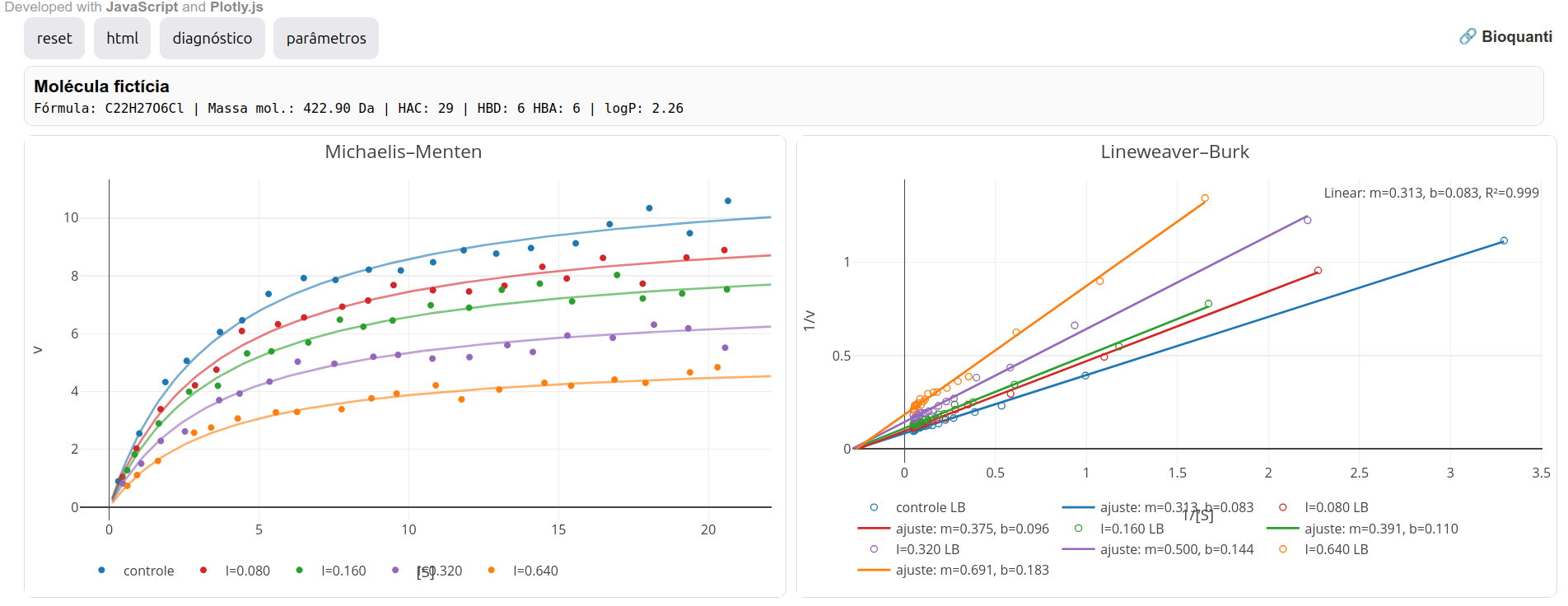
O código, um acrônimo para Lead-Hunt, atua como um aplicativo autônomo junto ao JSPlotly para simular a prospeção de composto bioativo como inibidor enzimático. O LH-Toolkit divide alguns conceitos e aplicações da catálise enzimática com o aplicativo visto na Seção 2.1, uma vez que envolve a visualização e interpretação de parâmetros e gráficos para cinética e inibição enzimáticas. Diferente desse, porém, tem como objetivo simular a prospeção para um composto líder por triagem (lead compound). A análise é realizada em função de comportamentos cinéticos e termodinâmicos para uma enzima potencialmente inibida por uma molécula fictícia. Como em todos os toolkits deste trabalho, a saída da simulação envolve um painel contendo parâmetros e gráficos usualmente utilizados na literatura pertinente, e acionados pelo botão adddo JSPlotly.

Diversos parâmetros cinéticos e termodinâmicos da interação do potencial inibidor com a enzima, bem como descritores da molécula fictícia, são simulados no tookit:
- Ki, constante de equilíbrio de dissociação do inibidor (desejável < 10\(\mu\)M);
- Vmax, velocidade limite da reação enzimática;
- Km, constante de Michaelis-Mentem;
- \(\Delta\)H, variação de entalpia de ligação com inibidor;
- \(\Delta\)S, variação de entropia de ligação com inibidor;
- \(\Delta\)Cp, variação de capacidade calorífica da ligação com inibidor;
Além desses parâmetros, a molécula fictícia informa alguns descritores, a saber:
- MW, peso molecular;
- HAC, número de átomos pesados (todos, exceto hidrogênio);
- HBD, número de doadores de ligação de H;
- HBA, número de aceptores de ligação de H;
- logP, logaritmo do coeficiente de partição da molécula
Esses descritores permitem o cálculo para outros critérios visando a seleção de líder, e descritos mais adiante no neste capítulo.
Onde: MW, kDa; faixas típicas pela regra de Lipinski (regra dos 5): MW<=500, HBD<=5, HBA<=10, cLogP<=5. E também LE >= 0.3; LLE >= 5.
A Regra de Lipinski ou regra dos cinco, reporta a um critério adotado pela Pfizer nos anos 1997 para auxiliar na triagem de novos candidatos a fármacos em seu estágio inicial de desenvolvimento. A regra baseia-se em quatro parâmetros físico-químicos, todos com valores múltiplos de cinco, para que um composto obtenha uma boa drogabilidade oral (drug-likeness).
Já a curva dose-reposta do inibidor permite a determinação de:
- IC\(_{50}\), concentração do inibidor que confere 50% de resposta (adequado quando < 10\(\mu\)M);
- nH, constante de Hill (índice de cooperatividade)
A figura abaixo ilustra as principais funcionalidades do toolkit, o qual pode ser baixado neste LINK do website Bioquanti.
3.2 Parâmetros
O LH-Toolkit permite trabalhar-se com diversos parâmetros cinéticos e termodinâmicos para interação do composto líder, a saber:
O tipo de inibição reversível (competitiva, incompetitiva, não competitiva pura, não competitiva mista);
O valor de Ki por construção de gráficos secundários variados a partir da tabela de preenchimento de parâmetros (Tabela para Ki) - plota-se a concentração I do inibidor versus Km, Vm, ou Km/Vm aparentes. Alternativamente, plota-se I contra o valor de (\(\alpha\)-1), onde \(\alpha\) representa “1+I/Ki”;
O valor de IC\(_{50}\) e de nH (inclinação do ponto médio) pela curva de dose-resposta do inibidor (estimativa de cooperatividade na ligação);
O valor de \(\Delta\)H e \(\Delta\)S para o plot de Van’t Hoff. No caso em que o gráfico apresentar-se curvilinear, também o valor valor de \(\Delta\)Cp;
Os valores de LE e LLE;
Avaliar o candidato junto às regras de Lipinski.
Dessa forma, pode-se utilizar o LeadHunt para uma simulação significativa à triagem de um inibidor como lead compound à uma enzima-alvo.
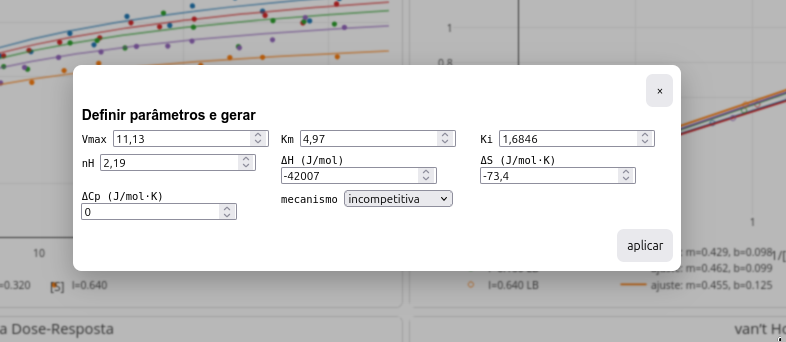
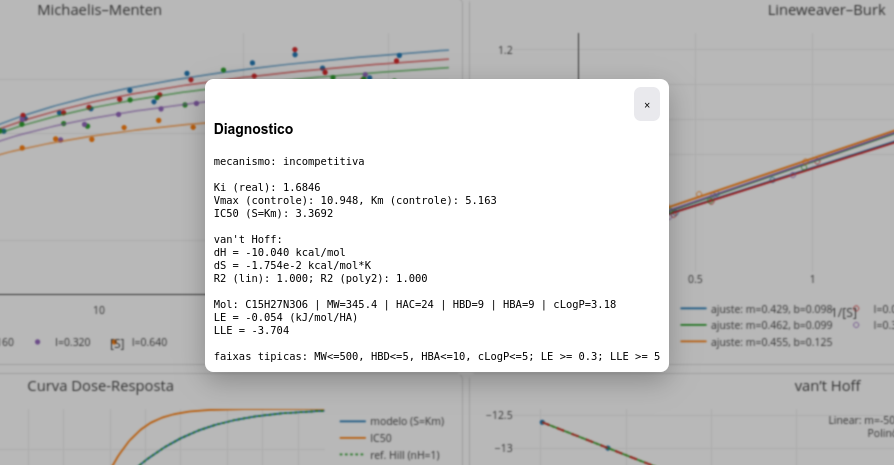
Há dois modos de estudo com uso do LeadHunt: inserindo-se parâmetros cinéticos e/ou termodinâmicos para observar os resultados expressos em gráficos (botão parâmetros), ou caracterizar os parâmetros a partir dos gráficos (botão diagnóstico).
No campo superior do app há 4 botões:
- reset - renova a simulação para outros dados e gráficos;
- html - salva os gráficos e dados em arquivo interativo;
- diagnóstico - abre janela popup para o diagnóstico do modelo de inibição e valores de parâmetros;
- parâmetros - abre janela para simulação gráfica frente a parâmetros do usuário.
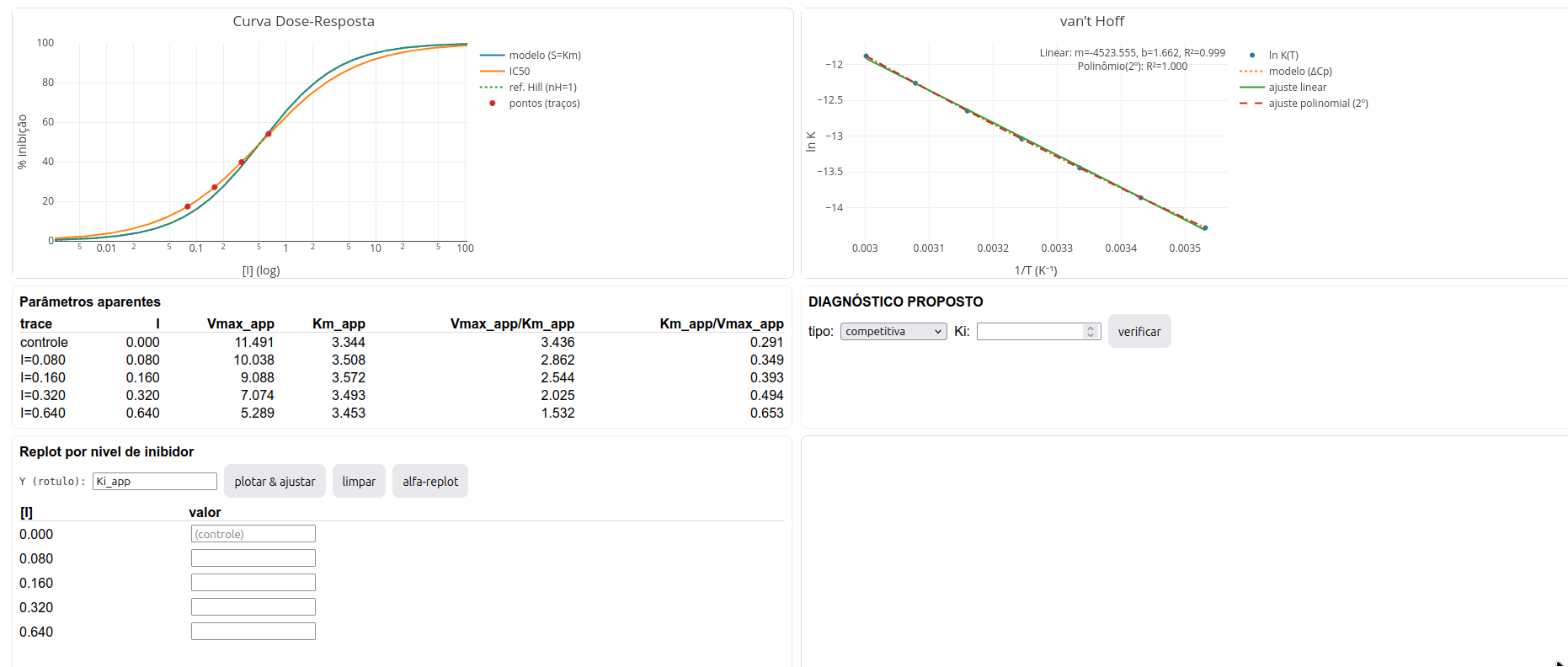
Os gráficos simulam os resultados obtidos para as representações de Michaelis-Mentem, Lineweaver-Burk, curva dose-reposta, e gráfico de van’t Hoff. As Figura 3.3 e Figura 3.4 ilustram as janelas para Parâmetros e Diagnóstico, respectivamente.
Para uma visão panorâmica do potencial de uso do toolkit, proceda como segue:
- Clique em reste para gerar um novo experimento (simulação, nova seed);
- Observe os gráficos de Michaelis-Mentem e duplor-recíproco gerados;
- Consulte a tabela de Parâmetros aparentes por nível de inibidor (opcionalmente, insira valores da tabela e elabore o gráfico (plotar & ajustar);
- Selecione um mecanismo de inibição em parâmetros;
- Observe a aba de curva dose-resposta na aba de %inibição contra o log(I);
- Observe os dois perfis nessa aba, um para a curva prevista quando S=Km (controle, nH=1), e a outra da simulação;
- Inspecione o gráfico de Van´t Hoff (1/T versus ln K) para o perfil linear ou curvilinear, como também para a inclinação positiva ou negativa;
- Abra a janela de diagnóstico e contraste suas observações com o esperado, bem como as faixas típicas para um composto líder;
- Exporte gráficos e resultados (html)
Se ficou confuso com essa panorâmica, sigua as atividades sugeridas para melhor apropriação do aplicativo junto aos conceitos de catálise e termodinâmica envolvidos.
3.2.1 Modelos de inibição
Diagnóstico do tipo (I x parâmetros)
Em paralelo às observações já estudadas na Seção 2.1 e espelhadas nos gráficos de Michaelis-Mentem e de Lineweaver-Burk no atual toolikit, é possível visualizar um modelo clássico de inibição por variação do teor de inibidor. Em poucas palavras, e partindo-se do pressuposto que um inibidor competitivo mantém constante o valor para Vmax, enquanto que um não competitivo mantém constante seu valor de Km, e um incompetitivo altera ambos de forma proporcional, pode-se pressupor algumas tendências gráficas, tal como observado na figura abaixo.
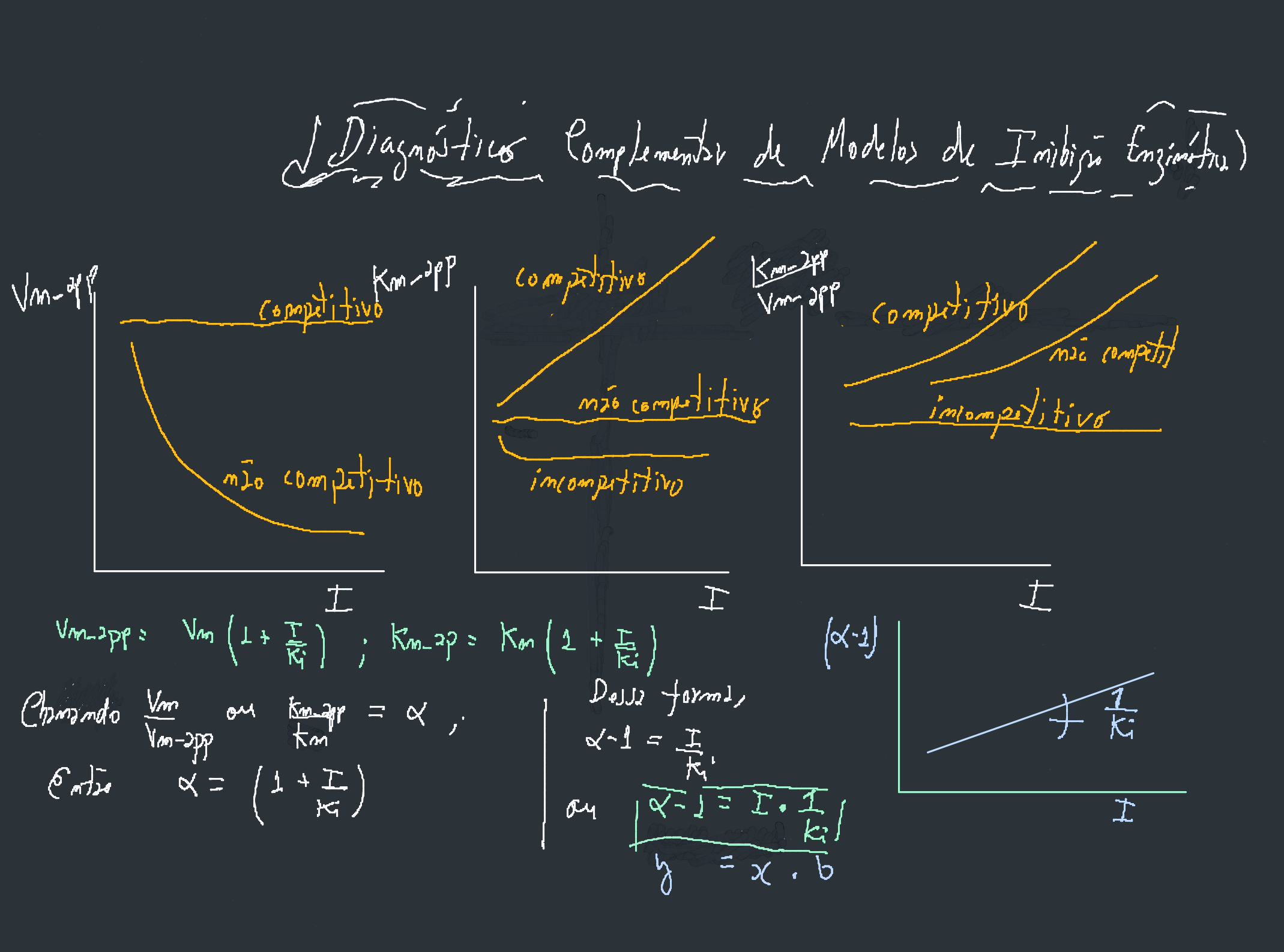
Pode-se utilizar o LH-Toolkit para validar essas afirmações. Para isso:
- Selecione um modelo qualquer: inibição competitiva, por exemplo (janela Parâmetros);
- Observe o resultado da simulação na janela Parâmetros aparentes;
- Copie e cole nos campos de valor os dados obtidos para Vmax_app (Km na presença do inibidor);
- Digite “Vmax_app” no campo de Y (rotulo), apenas para rotular o eixo dependente no gráfico;
- Clique em plotar & ajustar;
- Omita a linha de regressão linear da legenda, para visualizar somente os pontos.
A figura abaixo ilustra um resultado característico.
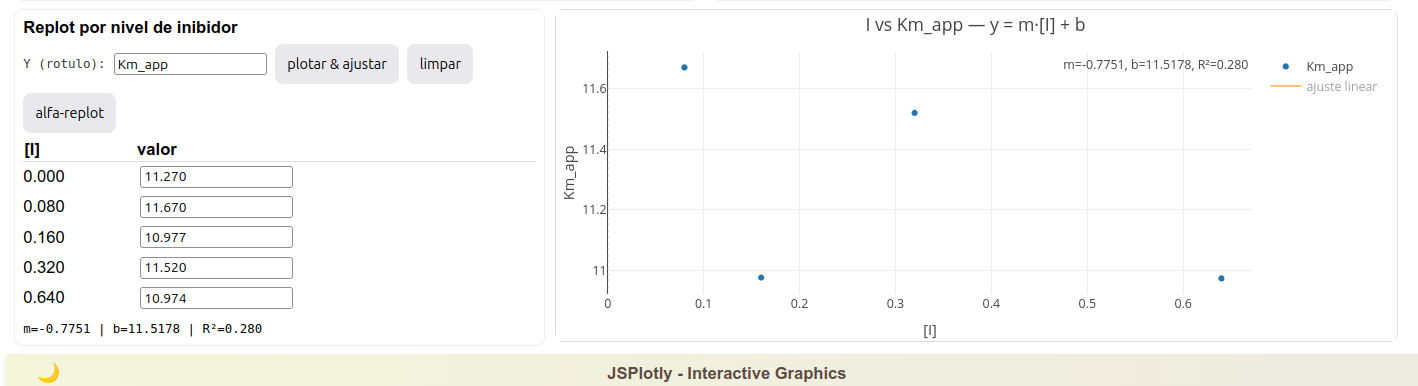
Observe que os pontos do gráfico de I versus Vmax_app parecem distribuir aleatoriamente os dados, sem uma tendência linear ou curvilinear clara. Esse resultado coaduna com o esperado, já que num modelo de inibição competitiva o valor de Vmax_app tende a permanecer constante, e com variação aleatória em função do erro relativo associado à dispersão.
Outros gráficos possíveis no replot, e com variações amplas de diagnóstico e quantificação, são realizados com Km_app, Km_app/Vmax_app, e Vmax_app/Km_app.
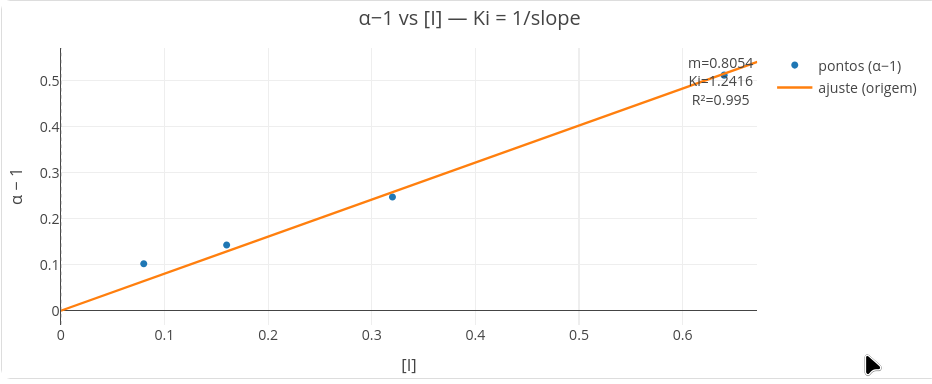
Determinação de Ki - I x (1-\(\alpha\))
Em paralelo à observação qualitativa para o tipo de inibição, também é possível a determinação da constante de dissociação Ki do complexo enzima-inibidor (em unidade \(\mu\)M). Esse valor pode ser obtido por replots secundários de Lineweaver-Burk, tal como apresentado na Seção 2.1. Neste toolkit, contudo, também é possível obter o parâmetro por uma estratégia simples, e deduzida na Figura 3.5. Em sínstese, um plot da concentração de inibidor contra o parâmetro (1-\(\alpha\)), e cuja inclinação fornece diretamente 1/Ki.
Para o LH-Toolkit, basta clicar em alfa replot, tal como representado abaixo.
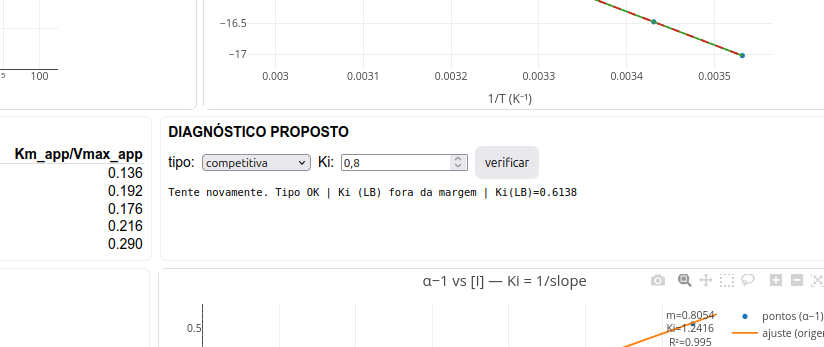
3.2.2 Diagnóstico pelo usuário
A fim de prover alguma atividade direta sobre o app de JSPlotly, o toolkit foi elaborado para permitir o diagnóstico do tipo de inibição e do valor de Ki apreciados pelo usuário. Para isso, basta inserir o tipo e valor nos campos adequados, tal com segue na figura abaixo. O algoritmo então valida ou não os valores inseridos em função dos parâmetros reais da simulação.
3.2.3 Eficiência de ligante - LE
O toolkit também simula valores para a LE e LLE, eficiência do ligante e eficiência lipofílica do ligante, respectivamente. Essas informações são utilizadas como descritores do potencial de ação do composto líder proposto. A eficiência do ligante é alternativamente definida pelas equações Equação 3.1 ou Equação 3.2:
\[ LE = 1.37 \frac{pIC_{50}}{HAC} \tag{3.1}\]
Onde pIC\(_{50}\) = -log(IC\(_{50}\)), e 1.37 refere-se a constante de conversão (RT * ln10, 25\(^{o}\)C).
\[ LE = \frac{\Delta G}{HAC} \tag{3.2}\]
Valores para LE podem ser interpretados como na Tabela 3.1 que segue.
| LE (kcal/mol/HA) | Interpretação |
|---|---|
| > 0.40 | Excelente - alta qualidade |
| 0.30 - 0.40 | Boa - adequada para otimização |
| 0.20 - 0.30 | Razoável - requer otimização |
| < 0.20 | Pobre - provavelmente não é bom líder |
Toolkit & LE
O aplicativo fornece na janela de Diagnóstico o valor obtido para LE, como observado na Figura 3.9.
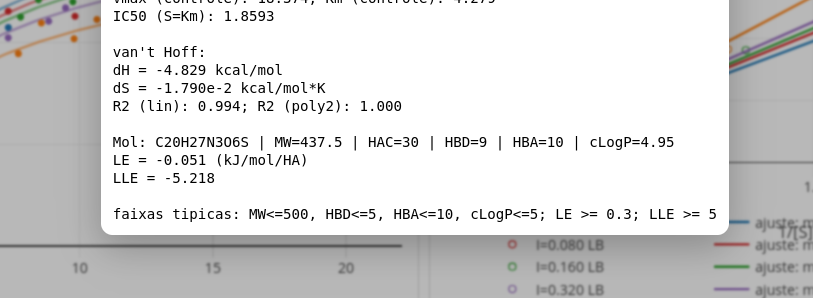
Perceba que o valor para LE está bem abaixo da faixa esperada, sugerindo uma molécula com baixa eficiência de ligação. Pela Equação 3.2, esse valor pode depender da variação de energia de ligação e/ou do quantitativo de átomos pesados da molécula. Perceba que a molécula fictícia exibe massa molecular limítrofe à regra dos 5, sugerindo que o baixo valor para LE seja devido mais à interação propriamente dita, do que ao ligante.
Para tornar mais claro a como analisar quantitativamente LE, segue um exemplo de cálculo para LE. Suponha que a molécula tenha a fórmula empírica C9H8O4 (aspirina), e um *IC\(_{50}\) de 100 nM. Nesse caso:
- o número de átomos pesados HAC é dado por 9C+ 40 = 13;
- pIC\(_{50}\) = -log(1x10\(^{-7}\)) = 7,0.
Dessa forma:
\[ LE=1.37*\frac{7}{13}=0.74 \]
Em suma, 0,74 kcal/mol por átomo pesado.
Um exemplo prático com LE
Suponha dois compostos inibidores da mesma enzima:
Composto A: - IC\(_{50}\) = 10 nM (pIC\(_{50}\) = 8.0) - 25 átomos pesados - LE = 1.37 × 8.0 / 25 = 0.44 kcal/mol/HA
Composto B: - IC\(_{50}\) = 5 nM (pIC\(_{50}\) = 8.3) - mais potente! - 40 átomos pesados - LE = 1.37 × 8.3 / 40 = 0.28 kcal/mol/HA
Conclusão: Embora B seja mais potente, A tem melhor eficiência e é provavelmente melhor candidato para otimização, pois usa menos átomos para obter boa potência.
3.2.4 Eficiência lipofílica do ligante - LLE
Outro descritor normalmente utilizado, semelhante ao parâmetro LE, é a eficiência lipofílica do ligante (lipophilic ligand efficiency - LLE). Esse parâmetro avalia a potência do composto, mas ajustada por sua lipofilicidade. Valores >5 são considerados bons.
\[ LLE=pIC_{50}-logP \tag{3.3}\]
3.2.5 Curva dose-resposta
As curvas dessa natureza mostram a tendência de resposta em função do teor de um desafiante (eficiência, morte, inibição, etc). Pode-se avaliar o perfil junto a ensaios tanto in vitro como in vivo. Assim, uma curva dose-resposta não está ligada ao fenômeno catalítico de enzimas propriamente dito, podendo extender-se em campo aberto a temas diversos, tais como células, roedores, extratos de plantas, etc. No LH-Toolkit essa curva pode é observada considerando uma molécula fictícia com alguma propriedades físico-químicas conhecidas sobre uma molécula enzimática. Dito dessa forma, a curva dose-resposta passa a agregar valor termodinâmico, podendo exibir parâmetros quantitativos expressos na representação de Hill, com aplicações em bioquímica, farmacodinâmica e toxicologia, tal como no esboço a seguir.
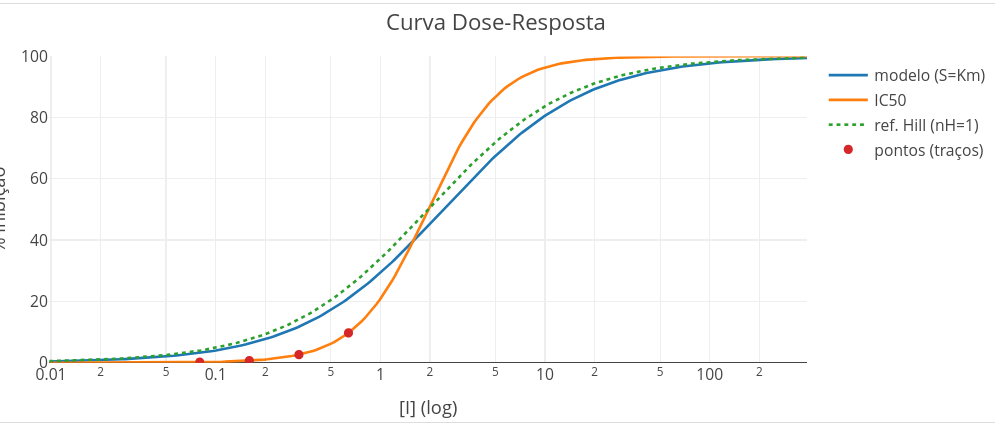
A relação utilizada para a construção do gráfico é dada Equação 3.4 abaixo.
\[ \%\,inh([I]) = \frac{100}{1 + \left(\frac{IC_{50}}{[I]}\right)^{n_H}} \tag{3.4}\]
O gráfico de Hill aborda a relação da quantidade complexo ligado/ligante livre sobre o logaritmo do teor do ligante livre (muito embora seja aceito na prática o valor de ligante total, considerando o livre < 5% desse). Para moléculas puras (ligante e macromolécula), o gráfico de Hill fornece parâmetros quantitativos, tais como Kd, a constante de equilíbrio de dissociação da molécula, bem como nH, o coeficiente de Hill. A magnitude de nH expressa a cooperatividade da interação do ligante, bem como a influência mútua dos sítios de ligação na macromolécula.
O LH-Toolkit apresenta a curva de Hill, sobrepondo o perfil previsto para nH unitário (sem cooperatividade) contra o perfil da simulação (exibindo ou não cooperatividade). Como representado (e bem mal escrito) na Figura 3.10, o valor de nH é menor do que a unidade para uma cooperatividade negativa entre sítios na macromolécula, e maior do que a unidade para uma cooperatividade positiva. Pelo formalismo clássico, contudo, o correto é tratar não por um sítio apenas, mas um conjunto de sítios, já que sítios de mesma afinidade passam desapercebido na análise de Hill, bem como em curvas de interação ligante-proteína. No LH-Toolkit, o resultado de simulação para o gráfico de Hill é representado na Figura 3.12.
Estrutura quaternária e nH
O valor de nH também se mostra útil no estudo de proteínas com mais de uma cadeia polipeptídica (quaternária), bem como para fenômenos de oligomerização e dissociação de monômeros. Nesse tipo de ensaio fixa-se a concentração de ligante enquanto se varia o teor de enzima. Se uma proteína se dissocia em monômeros ou sofre uma desnaturação (perda da estrutura quaternária em ambos), a cooperatividade geralmente desaparece, resultando em um valor de nH tendendo à unidade. Por outro lado, se monômeros sofrerem uma oligomerização em solução e houver interação positiva entre as cadeias, a interação do ligante exibirá um comportamento semelhante ao apresentado por estruturas quaternárias com cooperatividade positiva, ou seja, apresentando nH > 1. Agora, se num equilíbrio monômero-dímero o ligante só ligar ao monômero, o aumento da concentração da proteína favorecerá a formação do dímero, embora a interação com ligante só ocorra na forma monomérica da proteína. Nesse caso, o valor exibido para o coeficiente tenderá a nH < 1 (sequestro por oligomerização inativa). Isso também pode ocorre em situações em que o processo de oligomerização resulta na obstrução do sítio de interação para o ligante. A Figura 3.11 ilustra as situações mencionadas acima.
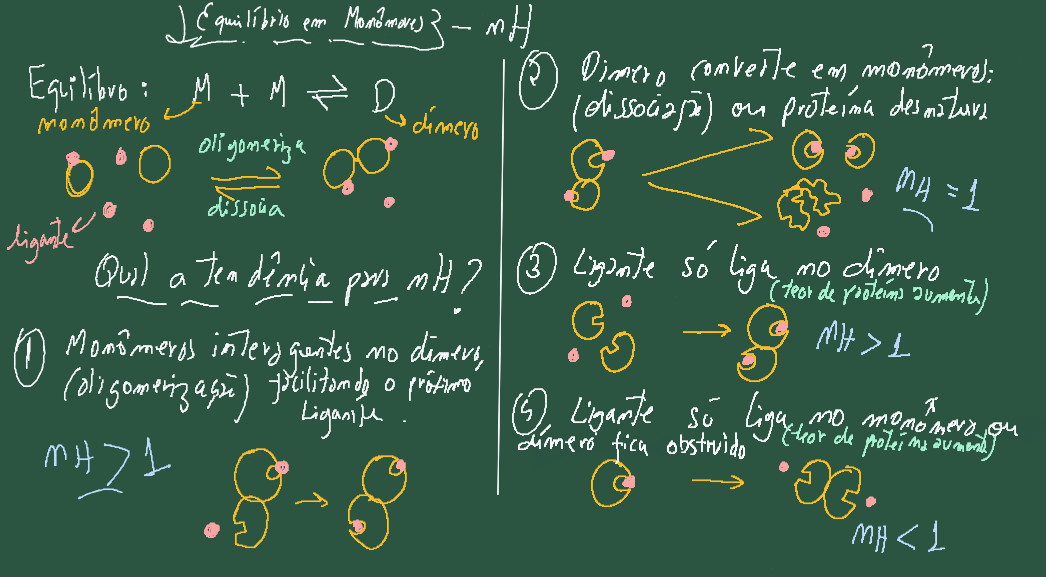
Assim, é possível estimar pelo toolkit se a interação da molécula candidata a composto líder dá-se por equilíbrio múltiplo de sítios, em paralelo a efeitos alostéricos mediados por transições conformacionais na enzima, e mesmo para fenômenos de oligomerização ou dissociação de estruturas quaternárias dessa. Daí o relativo mas abrangente poder diagnóstico do coeficiente de Hill.
A título de curiosidade…e confusão - IC\({50}\), Ki, e Kd !
O intercepto da curva logarítmica de Hill para um experimento de interação com ligantes é por vezes representado por log Kd. Da mesma forma, também o é para log IC\(_{50}\) para um ensaio de inibição enzimática, mas com uma ressalva bruta: não são parâmetros iguais, já que são extraídos de ensaios de natureza distinta. Isso decorre porque, embora IC\(_{50}\) e Kd ou Ki sejam definidos como teores de “algo” quando se tem 50% da resposta, IC\(_{50}\) também é determinado em condições experimentais em que a discussão termodinâmica perde validade, como nos ensaios com modelos animais e extratos de plantas, por exemplo.
3.2.6 Representação de van’t Hoff
Outro tratamento de dados utilizado para o diagnóstico da interação entre um inibidor e uma enzima, casos específicos da interação ligante-proteína, dá-se pelo gráfico de van’t Hoff apresentado pelo app na Figura 3.13 .
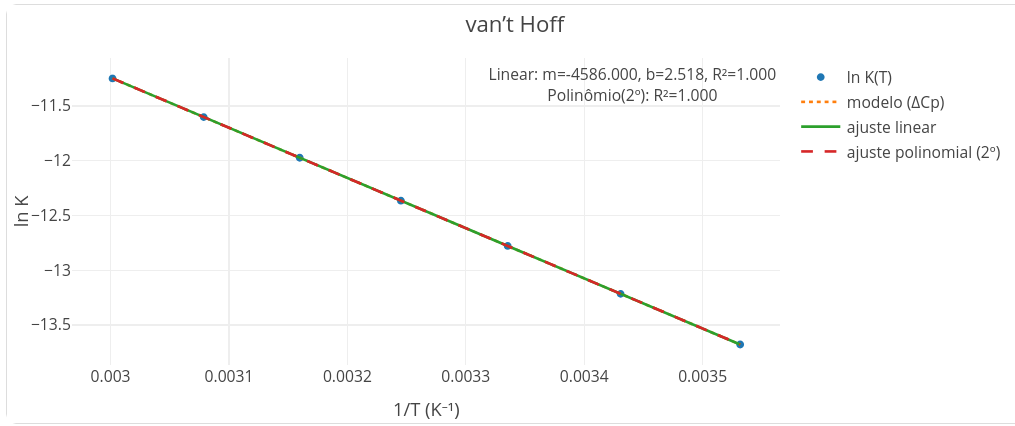
Essa representação é bastante diagnóstica, permitindo que se determine as quantidades termodinâmicas envolvidas na interação (\(\Delta\)H e \(\Delta\)S), bem como inferir sobre alterações conformacionais na estrutura da macromolécula sob interação (\(\Delta\)Cp). O quadro da Figura 3.14 ilustra um esboço para obtenção dessas quantidades.

A representação de van’t Hof exibe um duplo potencial para diagnóstico interacional, um em relação à inclinação do gráfico quando linear (Figura 3.13), e outro quando o gráfico apresenta-se curvilinear. Em relação à inclinação, é possível abstrair o sinal para a variação de entalpia \(\Delta\)H das equações que seguem, parte esboçada na Figura 3.14.
\[ \ln K = -\frac{\Delta H}{RT} + \frac{\Delta S}{R} \quad\Rightarrow\quad \text{slope} = -\frac{\Delta H}{R},\; \text{intercepto} = \frac{\Delta S}{R}. \tag{3.5}\]
Pela Equação 3.5, perceba que para um gráfico com inclinação positiva, o valor de \(\Delta\)H é negativo (Equação 3.6), o que equivale dizer que a interação é de característica exotérmica (ou exergônica). Por outro lado, se a inclinação linear for negativa, obrigatoriamente o valor para \(\Delta\)H é positivo, equivalente a uma interação de característica endotérmica (ou exergônica).
\[ \text{Inclinação} = \frac{d(\ln K)}{d(1/T)} \tag{3.6}\]
Para van´t Hoff curvilinear, contudo, não se abstrai a inclinação, mas o aspecto próprio da curva (Figura 3.14). A fuga de linearidade nessa representação indica que sua inclinação depende da temperatura. Isso equivale dizer que a variação da capacidade calorífica \(\Delta\)Cp do sistema não é nula, mas varia com a temperatura, o que resulta no aspecto curvilinear. Essa conclusão advém da definição própria para \(\Delta\)Cp:
\[ \Delta C_p = \left( \frac{\partial \Delta H}{\partial T} \right)_P \tag{3.7}\]
A Figura 3.15 ilustra essa curvatura para um \(\Delta\)Cp com valor negativo, tal como obtida pela janela de parâmetros do Toolkit.
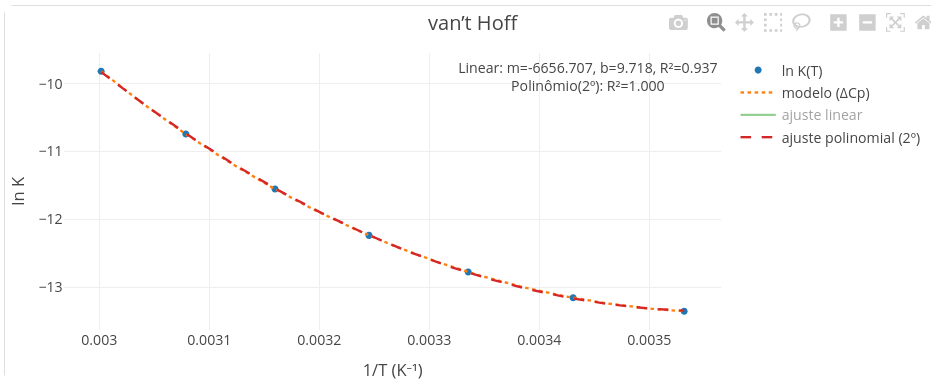
Perceba pelo gráfico acima que a inclinação aumenta com o aumento da temperatura, observada da direita para a esquerda, já o gráfico é recíproco nesse eixo. Como uma inclinação negativa em van’Hoff indica um um \(\Delta\)H positivo, então essa tangente sugere um \(\Delta\)H positivo que se eleva com a temperatura.
Pela Lei de Kirchhoff, se o valor de \(\Delta\)Cp é positivo, a entalpia da reação aumenta conforme a temperatura sobe. Ou seja, se a inclinação (tangente) é negativa, o \(\Delta\)H é positivo e, portanto, o processo absorve calor. Isso é usual quando a proteína sofre aumento da área de superfície por desnaturação em meio aquoso, sugerindo expansão de sua estrutura, um processo evidenciado na desnaturação térmica de proteínas, tal como monitorado por calorimetria de exploração diferencial - DSC. Valores negativos para \(\Delta\)Cp, por outro lado, sugerem compactação da estrutura, o que é comum em ligand-binding. Do ponto de vista mecanístico, esse processo usualmente ocorre com reorganização de moléculas de água por exclusão de solvente do sítio de interação (-\(\Delta\)S), acompanhada por interações específicas entre ligante e macromolécula (-\(\Delta\)H).
3.3 Outras características do toolkit
Assim como para o EK-Toolkit (Seção 2.1) e demais “caixas de ferramentas” desta obra, o LH-Toolkit possui algumas funcionalidades adicionais. Assim, o kit possibilita a exportação de um arquivo HTML autossuficiente contendo gráficos e diagnóstico de parâmetros (botão html). Os gráficos são interativos, permitindo zoom, linhas e pontos a apresentar/esconder por clique de mouse, e arrastes de eixos e plots.
Referências
- Marangoni, Alejandro G. Enzyme kinetics: a modern approach. John Wiley & Sons, 2003.
- Copeland, Robert A. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. John Wiley & Sons, 2013.
- Copeland, Robert A. Enzymes: a practical introduction to structure, mechanism, and data analysis. John Wiley & Sons, 2023.
- Leone, Francisco A. Fundamentos de cinética enzimática. Editora Appris, 2021.
- Ross, Philip D., and S. Subramanian. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry 20.11 (1981): 3096-3102.