Processos de adsorção entre ligantes e macromoléculas (proteína, ácido nucleico, glicano) - ligand bindig - envolvem a formação de um complexo cujas características auxiliam na definição de um possível candidato a composto líder (interações fármaco-receptor), no diagnóstico de biomarcadores, na detecção de alegenos em produtos alimentícios, no design de ligantes para controle de reatividade em metais, monitoramento de drogas, sistemas de liberação controlada, e mesmo ensaios de agregação celular, entre muitos exemplos.
As característica e os parâmetros advindos do complexo proteína-ligante (como de outros biopolímeros) são comumente determinados a partir de estudos in silico, por ancoragem molecular (docking) e dinâmica molecular, bem como de estudos in vitro, e esses com as mais diversas técnicas. Apenas para elencar alguns, a literatura reporta entre os estudos experimentais (Ligand Binding Assays - LBA) o emprego de ressonância magnética nuclear-RMN, difração de raios-X, dicroísmo circular, técnicas espectroscópicas (absorção molecular, fluorescência, infravermelho, ressonância de superfície plasmônica-SPR), métodos térmicos (calorimetria de titulação isotérmica-ITC, calorimetria de exploração diferencial-DSC), bem como hidrostáticas (viscosimetria, pressão osmótica) e piezoelétricas (microbalança de cristal de quartzo-QCM), para citar apenas algumas.
Não obstante o método empregado e as limitações de cada, as perguntas que se pretende responder acerca da interação entre um ligante e uma proteína são:
Quanto de proteína e de ligante existe ?
O complexo é formado ?
Quanto do complexo é formado ?
Quantos sítios existem para o ligante na macromolécula ?
Qual a força de interação para a formação do complexo ?
Quão rápido o complexo é formdo ?
Quais os mecanismos envolvidos na interação ?
Qual a força diretora da interação, entálpica ou entrópica ?
Quais as possíveis interações fracas envolvidas na formação do complexo ?
O que ocorre com a macromolécula sob interação com o ligante ?
O LB-Toolkit descrito neste capítulo pretende contribuir para responder àlgumas dessas questões, a partir de uma simulação em painel para ligand-binding, apresentando gráficos e parâmetros usualmente obtidos em ensaios in vitro.
O toolkit é um painel interativo para análise de ensaios de ligação ligante–receptor, permitindo gerar curvas, regressões clássicas (Scatchard, Benesi-Hildebrand, Hill) e análises termodinâmicas (Van’t Hoff). Ele simula, ajusta e interpreta parâmetros normalmente obtidos na experimentação, exibindo caixas com resultados numéricos, e oferecendo exportação HTML autônoma com todos os gráficos e valores.
4.1 Formação do complexo ligante-proteína
De modo geral, pode-se representar a interação ligante-proteína como segue:
Onde P representa o teor de proteína livre, L o ligante livre, e PL o complexo formado. As taxas de reação são definidas para a formação (k\(_{on}\); M\(^{-1}\)s\(^{-1}\)) e dissociação (k\(_{off}\); s\(^{-1}\)) do complexo.
Dessa forma deduz-se a equação para a isoterma de interação do ligante com a proteína como segue:
\[
Kd=\frac{[PL]}{[P]+[L]}
\tag{4.2}\]
Onde Kd representa a constante de equilíbrio de dissociação para o complexo PL formado, tal como condicionado ao equilíbrio de formação/dissociação do complexo (v\(_{assoc}\) = v\(_{dissoc}\)), e também definido como:
\[
Kd=\frac{k_{off}}{k_{on}}
\tag{4.3}\]
A partir da Equação 4.2 pode-se facilmente deduzir a expressão final para a interação de um ligante a um conjunto de sítios de mesma afinidade na proteína:
\[
\nu=\frac{n * [L]}{Kd + [L]}
\tag{4.4}\]
4.2 LB-Toolkit
O LB-Toolkit é um painel interativo para análise de ensaios de ligação ligante–receptor (proteína, enzima, ácido nucleico, glicano). Permite visualizar e ajustar curvas clássicas de ligação (Binding, Scatchard, Benesi–Hildebrand e Hill), e acompanha exportação HTML interativa de dados e gráficos.
O LB-Toolkit permite estudo e visualização gráfica de um painel 2 x 2 representativo dos modelos clássicos de interação bimolecular de Langmuir, Hill (cooperatividade positiva e negativa), heterogeneidade de sítios (two-site), e de transição conformacional (R/T). A exportação HTML é autossuficiente, e a importação/exportação para arquivos CSV dá-se por vetores L,v.
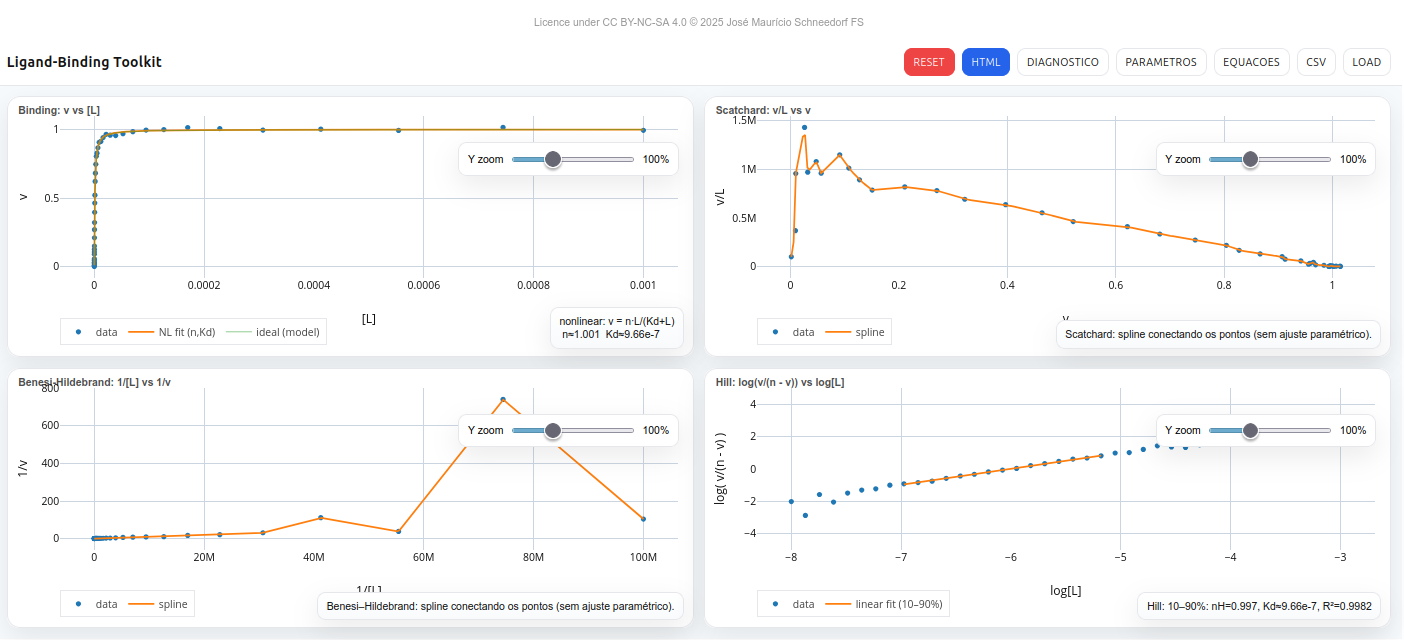
Figura 4.1: Tela do LB-Toolkit para interação macromolécula-ligante (ligand-binding). Acesse o script do aplicativo nesse LINK.
4.2.1 Funcionalidades
As funções principais da barra superior são: RESET (restaura valores padrão), HTML (exporta viewer autônomo), DIAGNOSTICO (mostra um resumo do modelo), PARAMETROS (abre painel de controle), EQUACOES (exibe fórmulas dos modelos), e CSV/LOAD (exporta ou importa dados em formato L,v.
Barra superior
Tabela de funcionalidades - LB-Toolkit
Botão
Função
RESET
Restaura valores padrão
HTML
Exporta viewer autônomo
DIAGNOSTICO
Mostra resumo do modelo
PARAMETROS
Abre painel de controle
EQUACOES
Exibe fórmulas em LaTeX
CSV / LOAD
Exporta ou importa dados (L,v)
Parâmetros ajustáveis
n, Kd, nH, erro sigma, points, Lmin, Lmax
Two-site: n1, n2, Kd1, Kd2
Conformational (R/T): Kt, KdR, KdT
Toggles: draw model only, results below, Hill 10–90%, zoom sliders, autoplot
Ajustes e estimativas
Binding (n,Kd): ajuste não linear (Gauss–Newton).
Scatchard e Benesi: ajuste linear.
Hill: ajuste linear em faixa de 10–90 % de ligante.
Gráficos
Binding: v vs [L] – curva teórica + pontos experimentais.
Scatchard: v/L vs v – linha spline ajustada aos dados.
Benesi–Hildebrand: 1/[L] vs 1/v – spline ilustrativo.
Hill: log(v/(n–v)) vs log[L] – regressão linear com exibição de nH e Kd.
Uso do LH-Toolkit
Para uma visão panorâmica do potencial de uso do toolkit, proceda como segue:
Observe os gráficos de Michaelis-Mentem e duplor-recíproco gerados;
Consulte a tabela de Parâmetros aparentes por nível de inibidor (opcionalmente, insira valores da tabela e elabore o gráfico (plotar & ajustar);
Selecione um mecanismo de inibição em parâmetros;
Observe a aba de curva dose-resposta na aba de %inibição contra o log(I);
Observe os dois perfis nessa aba, um para a curva prevista quando S=Km (controle, nH=1), e a outra da simulação;
Inspecione o gráfico de Van´t Hoff (1/T versus ln K) para o perfil linear ou curvilinear, como também para a inclinação positiva ou negativa;
Abra a janela de diagnóstico e contraste suas observações com o esperado, bem como as faixas típicas para um composto líder;
Exporte gráficos e resultados (html)
4.2.2 Modelos
Langmuir
\[
v = n\,\frac{[L]}{K_d + [L]}
\]
Hill
\[
v = n\,\frac{[L]^{n_H}}{K_d^{n_H} + [L]^{n_H}}
\]
Os ajustes são exibidos como linhas spline suaves, pois os parâmetros de uma regressão polinomial não têm significado físico direto.
Scatchard
\[
\frac{v}{[L]} = \frac{n}{K_d} - \frac{1}{K_d} v
\]
Benesi–Hildebrand
\[
\frac{1}{v} = a\,\frac{1}{[L]} + b
\]
Sua vez…
1. Utilize o "slider" (barra de deslizamento) para ampliação/redução dos gráficos;2. Experimente personalizar as represetações gráficas. Ilustrando, clique no botão "Parâmetros" e altere o modelo para cooperatividade negativa com dois sítios, atribuindo valores distintos a esses (opcional).
Agora você experimentar alguns cenários comuns da interação ligante-proteína encontrados na literatura.
Ligação simples: receptor se comporta como uma população homogênea. Para um receptor e uma “afinidade”, configure o toolkit para:
model ="standard"n =1.0Kd =1e-6nH =1.0err =0.02Lmin =1e-8Lmax =1e-3pts =40
Esse é o caso clássico de ligação reversível entre um ligante e uma macromolécula com sítios equivalentes e independentes. A curva v versus [L] deve ser hiperbólica: em baixa concentração de ligante, a ocupação cresce quase proporcionalmente, enquanto que em alta concentração, aproxima-se de um platô. O valor de Kd marca aproximadamente a concentração de ligante em que metade da capacidade máxima de ligação está ocupada.
Na curva direta, o platô se aproxima de n. No gráfico de Scatchard, o comportamento tende a ser aproximadamente linear para o caso ideal. No gráfico de Hill, o coeficiente angular esperado é próximo de 1, indicando ausência de cooperatividade forte.
Uma situação hipotética poderia envolver um Kd que diminui de 1e-6 para 1e-8. Nesse caso, observe que a curva se desloca para a esquerda. Isso significa que menos ligante é necessário para atingir a mesma ocupação, indicando que um menor Kd representa maior afinidade para a formação do complexo.
Mesmo receptor em dois ambientes físico-químicos. Para afinidade alta ve*rsus afinidade baixa, experimente:
Configuração (maior afinidade)
model ="standard"n =1.0Kd =1e-8err =0.02Lmin =1e-10Lmax =1e-4
Configuração B (menor afinidade)
model ="standard"n =1.0Kd =1e-5err =0.02Lmin =1e-8Lmax =1e-2
Esse cenário pode representar uma mesma proteína em dois ambientes distintos, como mudança de pH, força iônica, temperatura, mutação no sítio de ligação ou presença de moléculas que alteram a conformação da proteína. Em todos esses casos, a afinidade aparente pode mudar sem necessariamente mudar o número de sítios (conjunto).
Graficamente, a alteração de Kd desloca a curva no eixo de concentração. O platô permanece praticamente igual se n for mantido constante. Isso ajuda a separar dois conceitos frequentemente confundidos: capacidade máxima de ligação e afinidade.
Agora, você saberia responder se o Kd menor para um ligante indica que o composto, como um fármaco, por exemplo, é melhor ? Um menor Kd indica maior afinidade, embora eficácia biológica, seletividade, toxicidade, distribuição e cinética também sejam fatores importantes num estudo. O modelo desse capítulo descreve uma ligação em equilíbrio, e não o efeito fisiológico completo.
Cooperatividade positiva: o primeiro ligante abre caminho para os próximos. Uma ligação que facilita novas interações pode ser configurada abaixo:
model ="coop_pos"n =1.0Kd =1e-6nH =2.5err =0.02Lmin =1e-8Lmax =1e-3
Como visto em Seção 2.1, a cooperatividade positiva ocorre quando a ligação de uma molécula de ligante aumenta a probabilidade de outras ligações. A curva deixa de ser simplesmente hiperbólica e se torna mais sigmoidal. O gráfico de Hill foi historicamente usado para evidenciar esse tipo de comportamento, pois sua inclinação fornece uma estimativa operacional da cooperatividade. Em modelos de ligação, coeficientes de Hill maiores que 1 sugerem cooperatividade positiva, embora não devam ser confundidos automaticamente com o número real de sítios. (PMC)
No gráfico de Hill, a curva direta ganha uma transição mais abrupta. Ele deve apresentar inclinação maior que 1 na faixa intermediária de saturação. Na simulação do script, o ajuste de Hill é feito preferencialmente na faixa de 10–90%, mas apenas para facilitar a interpretação didática, já que essa faixa evita as distorções dos extremos da curva.
Você saberia dizer por que a região intermediária da curva é mais informativa do que os extremos ?
Nos extremos, a proteína está quase totalmente livre ou quase saturada. Pequenos erros experimentais podem gerar grandes distorções nas transformações logarítmicas. A faixa intermediária é mais sensível ao padrão de transição entre baixa e alta ocupação.
Cooperatividade negativa: afinidade diminui à medida que o sistema ocupa o primeiro ligante, dificultando os próximos. Uma configuração proposta para quando o primeiro ligante complexado dificulta a interação seguinte pode ser:
model ="coop_neg"n =1.0Kd =1e-6nH =0.55err =0.02Lmin =1e-8Lmax =1e-3
Também como observado em Seção 2.1, na cooperatividade negativa a ligação inicial reduz a afinidade dos sítios remanescentes, ou desloca o sistema para estados menos favoráveis à ligação. Em vez de uma transição abrupta, a curva pode parecer mais “espalhada” ao longo de várias ordens de grandeza de concentração.
O gráfico de Hill tende a apresentar inclinação menor que 1. A curva direta cresce de forma mais gradual, como se a população de macromoléculas não respondesse de maneira uniforme à concentração do ligante.
Então, conclui-se que a cooperatividade negativa significa que a proteína “não liga bem” ao ligante, correto ?
Não. Significa que a afinidade aparente muda durante o processo de ocupação. A proteína pode ligar fortemente o primeiro ligante, mas apresentar menor afinidade para eventos subsequentes.
Dois sítios independentes com afinidades diferentes. Para mais de um conjunto de sítios de ligação de afinidades distintas, experimente:
model ="two_site"n1 =0.5Kd1 =5e-8n2 =0.5Kd2 =2e-5err =0.015Lmin =1e-10Lmax =1e-2pts =60
Muitas proteínas não apresentam apenas um tipo de sítio de ligação. Pode haver dois sítios estruturalmente diferentes, duas populações conformacionais, heterogeneidade experimental ou mistura de isoformas. Neste cenário, o sistema total é a soma de duas contribuições, de uma alta afinidade por um lado, e de outra de baixa afinidade.
A curva direta pode parecer uma saturação alongada ou com dois regimes. O gráfico de Scatchard deixa de ser linear e passa a apresentar curvatura. Observe então que nem toda curvatura em Scatchard é automaticamente cooperatividade; ela também pode indicar heterogeneidade de sítios. Como distinguir cooperatividade real de mistura de dois sítios ? Somente pelo gráfico pode ser difícil. É preciso combinar modelo, controle experimental, ajuste quantitativo, dados estruturais e repetição com mutantes, competidores ou condições físico-químicas diferentes. Uma forma não experimental de conduzir essa distinção entre os dois modelos pode concretizar-se, por exemplo, por ajuste não-linear para cada equação (preferencialmente global, com todos os dados), seguido de teste de aderência dos parâmetros (goodeness-of-fit; Critério de Informação de Akaike, por ex).
Transição conformacional R/T: ligação como deslocamento de equilíbrio. Considerando possíveis efeitos alostérios no receptor:
Nesse modelo, R representa o estado relaxado, geralmente com maior afinidade pelo ligante, enquanto que T representa o estado tenso, geralmente com menor afinidade pelo ligante, o qual pode deslocar o equilíbrio entre esses estados.
Em geral, no modelo MWC clássico, \(K_{dR}\) possui valor menor que K\(_{dT}\), porque o estado R liga melhor o ligante do que o estado T. Já \(K_t\) representa o equilíbrio conformacional entre os estados T e R na ausência de ligante, influenciando mais quando a proteína está no estado T em relação ao estado R.
Uma leitura intuitiva da equação é:
========
[ v = n ]
O numerador:
[ L/K_{dR} + K_t L/K_{dT}]
soma as contribuições das formas ligadas:
[ RL]
e
[ TL]
O denominador:
[ 1 + L/K_{dR} + K_t(1 + L/K_{dT})]
soma todas as formas consideradas:
[ R]
[ RL]
[ T]
[ TL]
Ou seja:
[ = + + + ]
A sacada do MWC é que o ligante não apenas ocupa sítios: ele também muda a distribuição entre T e R. Se o ligante tem mais afinidade por R, conforme (L) aumenta, a proteína é “puxada” para o estado R, aumentando a ligação de modo cooperativo.
Em termos didáticos:
[ K_t ]
significa que a proteína começa mais “presa” no estado T, de baixa afinidade.
[ K_{dR} K_{dT}]
significa que o ligante prefere muito o estado R.
Juntos, esses fatores podem gerar uma curva sigmoidal: pouca ligação no começo, depois uma subida rápida, depois saturação.
=========================
conjunto de sítios mais apropriado que no. de sítios (vide herogen com 2 Kds mas 10 sítios, por ex)
Coop neg x Heterog: são parecidos (mas tem bimodal no hetero se exagerado Kd2)
Coop pos, hemoglobina, nH 2.8; Scatchard côncavo
MWC sem diff no Scatchard - comparar com hetero e coop.
MWC e hemoglob - modelo não MWC puro; mistura KNF
Van´t Hoff ausente: binding a T determinada não ventila diff conform e deltaCp>0, o qual só ocorre com diferentes T; além disso, binding não configura van´t Hoff curvo, normalmente
==========================
model ="conf_trans"n =1.0Kt =8.0KdR =5e-7KdT =2e-5err =0.02Lmin =1e-9Lmax =1e-3pts =60
Nessa situação ocorre uma transição conformacional entre estados R e T. A proteína já existe em equilíbrio entre conformações antes mesmo da ligação. O ligante se liga com afinidades diferentes a cada estado e, ao se ligar, desloca a população conformacional. Esse modelo alostérico também é conhecido por modelo MWC ou Monod-Wyman-Changeux, com proteínas alostéricas existindo em equilíbrio entre estados conformacionais, e ligantes estabilizando simultaneamente alguns estados em detrimento de outros (PMC).
No código do Toolkit, o modelo conformacional calcula a ocupação por uma expressão envolvendo Kt, KdR e KdT, permitindo comparar estados com afinidades distintas . Quando KdR é menor que KdT, o estado R apresenta maior afinidade, porque a Kd está representando uma constante de dissociação do complexo. Aumentar a constante de equilíbrio Kt, contudo, altera o peso relativo do estado T no equilíbrio e pode deslocar a curva de ligação.
Em resumo, Esose modelo descreve a alternância conformacinal da proteína entre dois estados, T e R. O ligante liga-se melhor ao estado R, e sua ligação desloca o equilíbrio conformacional, gerando cooperatividade. As constantes de dissociação K\(_{dR}\) e K\(_{dT}\) medem as afinidades em cada estado, enquanto K\(_t\) controla quanto a proteína favorece o estado T ou R antes da ligação.
O ligante “cria” o estado conformacional ativo ?
Não necessariamente. No modelo de seleção conformacional, os estados já existem em equilíbrio. O ligante estabiliza preferencialmente um deles, deslocando a distribuição populacional.
Ruído experimental e ilusão de mecanismo. Configuração sugerida:
model ="standard"n =1.0Kd =1e-6nH =1.0err =0.10Lmin =1e-8Lmax =1e-3pts =18
Depois compare com:
err =0.01pts =60
A curva direta pode ainda parecer razoável, mas os gráficos transformados ficam muito mais instáveis. Isso é particularmente visível em Benesi-Hildebrand, pois envolve 1/[L] e 1/v, ampliando erros quando L ou v são pequenos. O toolkit já exibe Benesi-Hildebrand como 1/[L] versus 1/v .
Transformações lineares como Scatchard, Benesi-Hildebrand e Hill são úteis para visualização, mas podem amplificar erros, especialmente em baixas concentrações, valores próximos de zero ou extremos de saturação. Isso ocorre porque a transformação matemática altera a distribuição dos erros. Pontos que eram apenas ruidosos podem ganhar peso exagerado no gráfico transformado. Por isso, a análise moderna tende a favorecer ajuste não linear direto aos dados originais, quando possível.
E se…
E se o mesmo conjunto de dados puder ser explicado por mais de um modelo ? Esse é um dos pontos mais importantes da Biofísico-química: ajustar uma curva não é o mesmo que provar um mecanismo. Uma curva sigmoidal pode sugerir cooperatividade, mas também pode surgir de mistura de populações, transições conformacionais ou limitações experimentais. O estudante deve aprender a perguntar não apenas “qual modelo ajusta melhor?”, mas “qual hipótese física esse modelo representa?”.
E se o gráfico de Scatchard for curvo? A curvatura pode indicar cooperatividade, heterogeneidade de sítios, mistura de proteínas, erro experimental ou faixa inadequada de concentração. O gráfico é uma pista, não uma sentença.
E se o coeficiente de Hill for 2,5 ? Isso não significa automaticamente que existem 2,5 sítios. O coeficiente de Hill é uma medida operacional da inclinação cooperativa na região intermediária da curva. Ele informa sobre o comportamento coletivo do sistema, não necessariamente sobre uma contagem estrutural direta.
E se o Kd estimado no ajuste direto não bater com o Kd do gráfico de Hill ? Isso pode acontecer porque cada método pesa os dados de maneira diferente. O ajuste direto trabalha sobre v versus [L]; o Hill transforma os dados em logaritmos e usa uma faixa específica de saturação. A divergência pode ser uma oportunidade didática para discutir erro, modelo e inferência.
Outras características do toolkit
Assim como para o EK-Toolkit (Seção 2.1) e demais “caixas de ferramentas” desta obra, o LH-Toolkit possui algumas funcionalidades adicionais. Assim, o kit possibilita a exportação de um arquivo HTML autossuficiente contendo gráficos e diagnóstico de parâmetros (botão html). Os gráficos são interativos, permitindo zoom, linhas e pontos a apresentar/esconder por clique de mouse, e arrastes de eixos e plots.
Referências
Canals, Meritxell et al. A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. Journal of Biological Chemistry, v. 287, n. 1, p. 650-659, 2012.
Carey, Jannette. Ligand-Binding Basics: Evaluating Intermolecular Affinity, Specificity, Stoichiometry, and Cooperativity. John Wiley & Sons, 2025.
Mannhold, Raimund, Hugo Kubinyi, and Gerd Folkers. Protein-ligand interactions: from molecular recognition to drug design. John Wiley & Sons, 2006.
Klotz, Irving. Introduction to biomolecular energetics: including ligand–receptor interactions. Elsevier, 2012.
Roque, Ana Cecília A. “Ligand-Macromolecular interactions in drug discovery.” Clifton, NJ 2010; p 572 (2010).